Pituitary microadenoma, macroadenoma and nonfunctioning adenomas (NFPAs), hyperprolactinemia syndrome
- Pituitary Adenoma (Microadenoma, Macroadenoma), Hyperprolactinemia Syndrome Overview
- Pituitary Microadenomas:
- Pituitary Macroadenomas:
- Nonfunctioning Pituitary Adenomas (NFPAs)
- Pituitary Adenoma and Hyperprolactinemia Syndrome Diagnosis
- Pituitary Adenoma and Hyperprolactinemia Syndrome Treatment
- Treatment of Nonfunctioning Pituitary Adenomas (NFPAs)
- References
Pituitary Adenoma (Microadenoma, Macroadenoma), Hyperprolactinemia Syndrome Overview
Pituitary adenomas are typically benign (non-cancerous) tumors originating from the hormone-producing cells of the anterior pituitary gland (adenohypophysis), located at the base of the brain within a bony structure called the sella turcica [1]. These adenomas are classified based on size (microadenoma versus macroadenoma) and hormonal activity (functioning versus nonfunctioning).
Hyperprolactinemia syndrome refers to the clinical consequences of persistently elevated levels of prolactin, a hormone normally involved in lactation. Prolactinomas (prolactin-secreting pituitary adenomas) are the most common type of functioning pituitary adenoma and a frequent cause of hyperprolactinemia [2]. In women, symptoms typically include galactorrhea (inappropriate milk production), menstrual irregularities (oligomenorrhea or amenorrhea), and infertility. In men, hyperprolactinemia often leads to decreased libido, erectile dysfunction, infertility due to oligospermia, gynecomastia (breast enlargement), and less frequently, galactorrhea. Elevated prolactin inhibits gonadotropin-releasing hormone (GnRH) secretion from the hypothalamus, leading to secondary hypogonadism.

Symptoms related to pituitary adenomas arise either from excess hormone secretion (in functioning adenomas) or from the tumor's physical size and pressure on surrounding structures (mass effect), particularly with macroadenomas. Hormone excess syndromes include:
Excess Hormone |
Resulting Condition & Key Health Problems |
|---|---|
| Prolactin (PRL) |
Hyperprolactinemia: - Females: Menstrual dysfunction (oligomenorrhea, amenorrhea), galactorrhea, infertility, decreased bone density. - Males: Decreased libido, erectile dysfunction, infertility, gynecomastia (less common), decreased bone density. [2] |
| Growth Hormone (GH) |
Acromegaly (adults) / Gigantism (children): - Enlargement of hands, feet, facial features (jaw, brow). - Soft tissue swelling, excessive sweating. - Osteoarthritis, carpal tunnel syndrome. - Sleep apnea. - Hypertension, cardiomyopathy. - Diabetes mellitus or impaired glucose tolerance. - Increased risk of colorectal polyps/cancer. [3] |
| Adrenocorticotropic Hormone (ACTH) |
Cushing's Disease (Pituitary-dependent Cushing's Syndrome): - Central obesity (truncal fat, moon facies, buffalo hump), thin skin, easy bruising, purple striae. - Muscle weakness (proximal myopathy), osteoporosis. - Mood changes (anxiety, depression, psychosis). - Hypertension, diabetes mellitus. - Increased susceptibility to infections. - Menstrual irregularities (females). [4] |
| Thyroid-Stimulating Hormone (TSH) |
TSH-oma (rare): - Secondary Hyperthyroidism: Symptoms include heat intolerance, sweating, tachycardia/palpitations (arrhythmias like atrial fibrillation), tremor, anxiety/irritability, unintentional weight loss despite increased appetite, goiter. Characterized by inappropriately normal or elevated TSH with elevated free T4/T3 levels. [5] |
| Gonadotropins (LH, FSH) |
Gonadotroph Adenoma (often clinically nonfunctioning): - Usually secrete inactive or intact hormone fragments (e.g., alpha-subunit, beta-FSH, less commonly intact FSH/LH). - May rarely cause ovarian hyperstimulation (females) or testicular enlargement/precocious puberty (males). - Most often present due to mass effect or are discovered incidentally. [6] |
Nonfunctioning pituitary adenomas (NFPAs) do not secrete clinically significant amounts of active hormones, although many produce hormone subunits detectable by special tests. They typically cause symptoms due to mass effect when they become macroadenomas, compressing adjacent structures like the optic chiasm (causing visual field defects, classically bitemporal hemianopia), cranial nerves within the cavernous sinus (causing double vision), or normal pituitary tissue (causing hypopituitarism - deficiency of one or more pituitary hormones). Headaches are also common due to dural stretching or increased sellar pressure.
Pituitary Microadenomas
Description of Pituitary Microadenomas
A pituitary microadenoma is defined as a pituitary adenoma with a maximum diameter of less than 10 millimeters (less than 1 cm) [7]. These tumors are confined within the sella turcica and typically do not cause mass effect symptoms like visual loss. Microadenomas can be either functioning (hormone-secreting) or nonfunctioning. Functioning microadenomas cause symptoms related to the specific hormone they overproduce (e.g., prolactinoma causing hyperprolactinemia, ACTH-secreting causing Cushing's disease, GH-secreting causing acromegaly).
Many microadenomas, however, are clinically nonfunctioning and are discovered incidentally during brain imaging (Magnetic Resonance Imaging - MRI or CT) performed for other reasons (e.g., headache, dizziness, trauma). These are often referred to as pituitary 'incidentalomas'. While the vast majority are benign findings requiring only monitoring, even small incidental microadenomas can sometimes cause subtle hormonal imbalances (subclinical Cushing's, mild hyperprolactinemia) or, rarely (< 10% over several years), grow over time [8].
MRI with gadolinium contrast, particularly using dynamic contrast-enhanced sequences and thin slices through the pituitary fossa, is the imaging modality of choice for detecting microadenomas. They often appear as small, focal lesions that enhance less avidly or later than the surrounding normal pituitary gland tissue (typically hypointense on early post-contrast T1 images) [9].
Clinically significant microadenomas are most commonly prolactinomas, followed by ACTH-secreting adenomas causing Cushing's disease, and GH-secreting adenomas causing acromegaly. Nonfunctioning microadenomas are often gonadotroph adenomas by immunohistochemistry but lack clinical hormone excess. Differential diagnosis on imaging includes Rathke's cleft cysts, pituitary hyperplasia, metastases, or inflammatory processes (hypophysitis), although these often have distinct characteristics or clinical context.
Pathophysiology of Pituitary Microadenomas
Pituitary adenomas, including microadenomas, are generally considered monoclonal neoplasms, meaning they arise from the clonal expansion of a single mutated anterior pituitary cell [10]. The specific initiating mutations and subsequent genetic/epigenetic events leading to adenoma formation are complex and varied, but several factors and pathways are implicated.
Specific germline genetic syndromes predispose individuals to pituitary adenomas: Multiple Endocrine Neoplasia type 1 (MEN1), caused by mutations in the MEN1 tumor suppressor gene, frequently involves prolactinomas or GH-secreting adenomas. McCune-Albright syndrome, due to mosaic activating mutations in the GNAS gene encoding the Gs-alpha protein subunit, often leads to GH-secreting adenomas (somatotropinomas). Carney complex (PRKAR1A mutations) can include pituitary adenomas (typically GH-secreting). Mutations in the AIP (aryl hydrocarbon receptor interacting protein) gene predispose to familial isolated pituitary adenomas (FIPA), particularly GH-secreting or prolactin-secreting tumors, often presenting at a young age and being larger/more aggressive [11].
Sporadic (non-familial) adenomas also involve somatic genetic and epigenetic alterations. Activating somatic mutations in GNAS are found in about 40 percent of sporadic GH-secreting adenomas. Mutations in USP8 are common in ACTH-secreting adenomas [12]. Other somatic mutations and epigenetic changes affecting cell cycle regulators (e.g., loss of function in CDKN1B/p27, alterations in p53, RB pathways), growth factor pathways (e.g., overexpression of FGFR4), and pituitary-specific transcription factors contribute to tumorigenesis and hormone production. Loss of heterozygosity at certain chromosomal locations (e.g., 11q13 where MEN1 resides) is also observed.
Prolactinomas, the most common type, arise from lactotroph cells. Corticotropinomas causing Cushing's disease arise from corticotroph cells. Somatotropinomas causing acromegaly arise from somatotroph cells. Gonadotropinomas usually arise from gonadotroph cells and often secrete inactive hormone subunits (alpha-subunit, beta-FSH, beta-LH). Thyrotropinomas causing hyperthyroidism arise from thyrotroph cells and are rare.
Most clinically nonfunctioning adenomas are immunopositive for gonadotropin subunits (FSH, LH, alpha-subunit) but do not cause clinical syndromes of hormone excess. Some may be "null cell" adenomas with no specific hormone staining, or silent corticotroph/somatotroph adenomas.
Epidemiology of Pituitary Microadenomas
Frequency of Pituitary Microadenomas
Pituitary microadenomas are surprisingly common incidental findings. Autopsy studies have reported microscopic or small adenomas (often < 3mm) in up to 27 percent of individuals without known pituitary disease [13, 14]. High-resolution imaging studies (MRI) detect pituitary incidentalomas (most of which are microadenomas < 10mm) in approximately 10-20 percent of the general population undergoing brain scans for unrelated reasons [15].
The vast majority of these incidentally discovered microadenomas are nonfunctioning and remain stable or grow very slowly, never causing clinical symptoms. Functioning microadenomas are much less common but are the usual cause of pituitary hormone excess syndromes like hyperprolactinemia (prolactinoma being the most frequent clinically diagnosed type, estimated prevalence ~10-50 per 100,000 population [16]), Cushing's disease, and acromegaly.
Pituitary microadenomas can occur at any age but are most commonly diagnosed in young to middle-aged adults (20s-50s). There is a female predominance for prolactinomas (F:M ratio ~10:1) and Cushing's disease (~3-5:1), while GH-secreting adenomas have roughly equal gender distribution.
Before widespread MRI use, hyperprolactinemia detected via blood tests was a major clue to underlying, often unseen, prolactin-secreting microadenomas. MRI now allows direct visualization.
The high prevalence of microadenomas found incidentally compared to the lower prevalence of clinically significant pituitary disease or macroadenomas suggests that most microadenomas do not progress to become large or symptomatic tumors.
Pituitary Macroadenomas
Description of Pituitary Macroadenomas
A pituitary macroadenoma is defined as a pituitary adenoma measuring 10 millimeters (1 cm) or greater in maximum diameter [3]. Unlike microadenomas, macroadenomas frequently extend beyond the confines of the sella turcica, potentially compressing surrounding neural and vascular structures.
They can be either functioning (hormone-secreting) or nonfunctioning. Nonfunctioning pituitary adenomas (NFPAs) are the most common type of macroadenoma diagnosed clinically [6]. Functioning macroadenomas include large prolactinomas, GH-secreting adenomas causing acromegaly, and less commonly, large ACTH-secreting adenomas (often associated with Nelson's syndrome after adrenalectomy) or TSH-secreting adenomas.
Macroadenomas often present with symptoms related to mass effect due to their size:
- Visual Disturbances: Compression of the optic chiasm located superior to the pituitary is common with suprasellar extension, classically causing bitemporal hemianopia (loss of peripheral vision in both outer temporal fields). Compression of optic nerves or tracts can cause other patterns of vision loss or reduced visual acuity.
- Cranial Nerve Palsies: Lateral extension into the cavernous sinus can compress cranial nerves III (oculomotor), IV (trochlear), V (trigeminal - V1 ophthalmic and V2 maxillary branches), and VI (abducens), leading to double vision (diplopia), eyelid drooping (ptosis), pupillary changes, or facial numbness/pain. CN III and VI are most commonly affected clinically.
- Headaches: Often nonspecific, potentially due to stretching of the dura mater covering the sella (diaphragma sellae) or increased intrasellar pressure.
- Hypopituitarism: Compression and destruction of the normal pituitary gland tissue can lead to deficiency of one or more anterior pituitary hormones (GH deficiency is often earliest, followed by gonadotropins LH/FSH causing hypogonadism, then TSH causing secondary hypothyroidism, and lastly ACTH causing secondary adrenal insufficiency) or, less commonly, posterior pituitary dysfunction (Antidiuretic Hormone - ADH deficiency causing central diabetes insipidus, more often seen with stalk compression or lesions originating posteriorly).
- Pituitary Apoplexy: An acute medical emergency caused by sudden hemorrhage or infarction within a pituitary adenoma (much more common in macroadenomas than microadenomas). Presents with sudden severe headache, rapid vision loss, cranial nerve palsies, altered consciousness, and often acute hypopituitarism (requiring urgent steroid replacement).
Functioning macroadenomas also cause symptoms related to hormone overproduction, often more severe than with microadenomas due to larger tumor burden, as described earlier.
Pathophysiology of Pituitary Macroadenomas
The underlying pathophysiology involves the clonal expansion of mutated adenohypophyseal cells, similar to microadenomas. The factors driving progression from microadenoma to macroadenoma or the de novo development of macroadenomas are not fully elucidated but likely involve accumulation of additional genetic (e.g., mutations in tumor suppressor genes like p53, chromosomal instability) or epigenetic alterations promoting increased proliferation, decreased apoptosis, angiogenesis, and sometimes invasiveness [10].
Macroadenomas often demonstrate loss of normal feedback mechanisms controlling hormone secretion (especially functioning ones). Their growth leads to compression and destruction of adjacent normal pituitary cells, resulting in hypopituitarism. Extension superiorly compresses the optic chiasm and potentially the hypothalamus (leading to DI or appetite/temperature dysregulation). Lateral extension invades the cavernous sinus, potentially encasing the internal carotid artery and cranial nerves III, IV, V1, V2, and VI. Inferior extension can erode the floor of the sella turcica into the sphenoid sinus, sometimes causing CSF rhinorrhea.
The genetic basis involves factors mentioned for microadenomas, including germline syndromes (MEN1, GNAS, AIP, Carney complex) and somatic mutations (GNAS, USP8). Additional alterations associated with larger or more aggressive tumors might include mutations in cell cycle regulators (p53, RB), growth factor receptors (FGFR4), or adhesion molecules. While benign histologically (low mitotic rate, no cytologic atypia usually), some macroadenomas exhibit invasive growth patterns into surrounding structures (dura, bone, cavernous sinus), making complete surgical resection challenging. True primary pituitary carcinomas, defined by the presence of craniospinal or systemic metastases, are exceedingly rare.
Associated clinical syndromes like MEN1, McCune-Albright, and Carney complex can involve macroadenomas, often GH-secreting or prolactin-secreting types.
Epidemiology of Pituitary Macroadenomas
Frequency of Pituitary Macroadenomas
While incidental microadenomas are very common, clinically significant pituitary tumors requiring evaluation and potential treatment are less frequent. The overall prevalence of clinically apparent pituitary adenomas is estimated at around 78-94 per 100,000 individuals, or roughly 1 in 1000 [17, 18]. Macroadenomas constitute a significant proportion of these clinically relevant tumors, likely representing 30-50% of diagnosed adenomas, particularly among nonfunctioning adenomas which often present due to size.
Population-based studies suggest an overall annual incidence of newly diagnosed pituitary adenomas around 4 per 100,000 population [18], with prevalence rates increasing due to improved detection with imaging. The incidence requiring neurosurgical intervention ranges from approximately 1 to 7 per 100,000 per year, with macroadenomas causing mass effect or hormone excess refractory to medical therapy being the primary indications for surgery.
Morbidity and Mortality in Pituitary Macroadenomas
Morbidity associated with pituitary macroadenomas stems primarily from mass effects (visual loss, cranial nerve palsies, debilitating headaches), hormonal dysfunction (hypopituitarism requiring lifelong replacement, or the systemic consequences of hormone excess syndromes like acromegaly or Cushing's disease), and potential complications of treatment (e.g., surgical risks like CSF leak, meningitis, vascular injury, worsening hypopituitarism; long-term effects of radiation like hypopituitarism, secondary tumors, cognitive changes). Visual impairment, particularly permanent deficits from delayed treatment of chiasmal compression, significantly impacts quality of life. Hypopituitarism requires careful management and carries its own risks if inadequately treated (e.g., adrenal crisis). Complications of uncontrolled hormone excess (e.g., cardiovascular disease, diabetes, osteoporosis, increased cancer risk in acromegaly and Cushing's) contribute significantly to long-term morbidity and mortality if not effectively managed [19, 20].
Mortality directly related to the benign nature of pituitary macroadenomas themselves is generally low. However, increased mortality risk is well-documented in patients with uncontrolled acromegaly and Cushing's disease, primarily due to cardiovascular complications (hypertension, heart failure, stroke, arrhythmias), metabolic issues (diabetes), and infections (Cushing's). Pituitary apoplexy, while rare, can be fatal if not recognized and treated promptly. Complications from surgery (though mortality is low, <1%, in experienced centers) or long-term effects of radiation can also contribute. Overall survival for patients with macroadenomas is generally good with modern multidisciplinary management that controls hormone levels and mass effect, although quality of life may be affected by residual symptoms or treatment side effects.
Pituitary macroadenomas show no significant racial predilection.
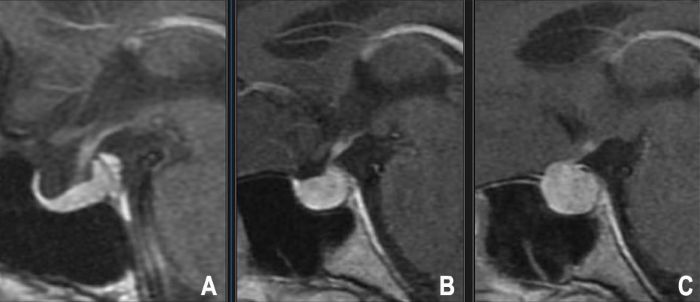
While autopsy studies show equal prevalence of incidental adenomas between sexes, clinically diagnosed prolactinomas and ACTH-secreting adenomas (Cushing's disease) are significantly more common in women of reproductive age, possibly due to earlier symptom recognition (menstrual irregularities, galactorrhea). GH-secreting adenomas (acromegaly) and clinically presenting NFPAs have a more equal gender distribution. Pituitary macroadenomas can occur at any age but are most frequently diagnosed in adults, with incidence peaking between the third and sixth decades.
Nonfunctioning Pituitary Adenomas (NFPAs)
Nonfunctioning pituitary adenomas (NFPAs) are pituitary tumors that do not cause a clinical syndrome of hormone over-secretion. They represent approximately 15-30 percent of all pituitary adenomas diagnosed clinically and are the most common type presenting as macroadenomas [22]. While termed "nonfunctioning," immunohistochemical staining reveals that most NFPAs originate from gonadotroph cells and often produce gonadotropin hormones (Follicle-Stimulating Hormone - FSH, Luteinizing Hormone - LH) or their subunits (particularly the alpha-subunit or FSH-beta subunit), but these hormones are typically biologically inactive, secreted inefficiently, or secreted in abnormal ratios, thus not causing clinical symptoms of hormone excess [6]. Some may be true "null cell" adenomas or silent variants of other hormone-producing cells.
Because they lack hormone hypersecretion syndromes, NFPAs usually present later than functioning adenomas, often when they have grown large enough (macroadenomas) to cause symptoms due to mass effect:
- Visual field defects (typically bitemporal hemianopia from optic chiasm compression).
- Headaches (nonspecific, due to dural stretch or sellar pressure).
- Hypopituitarism (due to compression of normal pituitary tissue, most commonly affecting GH and gonadotropin secretion first, followed by TSH and ACTH).
- Cranial nerve palsies (less common, from cavernous sinus invasion, causing diplopia or facial numbness).
- Pituitary apoplexy (acute hemorrhage/infarction within the tumor).
Many smaller NFPAs are discovered incidentally (MRI or CT) as pituitary incidentalomas. Autopsy and imaging studies suggest a high prevalence of silent microadenomas (up to 1 in 6 individuals potentially harboring one), but most remain small and asymptomatic throughout life.
The primary treatment for symptomatic NFPAs causing mass effect or progressive hypopituitarism is surgical resection, usually via the transsphenoidal route. The main goals are decompression of surrounding structures (especially the optic apparatus to improve/preserve vision) and maximal safe tumor removal to reduce mass effect and potentially improve pituitary function. Management of asymptomatic incidental NFPAs involves monitoring with serial imaging and endocrine/visual assessments [23].
Pituitary Adenoma and Hyperprolactinemia Syndrome Diagnosis
Differential Diagnosis of Sellar/Suprasellar Masses
| Condition | Key Features / Distinguishing Points | Typical Imaging Findings (MRI) |
|---|---|---|
| Pituitary Adenoma (Micro/Macro) | Endocrine symptoms (excess/deficiency) or mass effect (vision loss, headache, CN palsy). Originates within sella, displaces gland, may extend supra/parasellar. | Micro: Focal lesion <10mm, typically T1 hypointense, enhances less/later than normal gland. Macro: Sellar mass ≥10mm, variable enhancement (often less than gland), may show hemorrhage/cyst, extends supra/parasellar, may erode bone. |
| Craniopharyngioma | Bimodal age peak (children/older adults). Suprasellar location common. Headaches, visual loss, endocrine dysfunction (GH def, DI common), hydrocephalus. | Often mixed cystic (T1 variable/T2 hyper) and solid components. Calcification very common (CT best). Enhances (solid parts, thick cyst wall). Typically suprasellar epicenter, may adhere to surrounding structures. |
| Meningioma (Sellar/Suprasellar/Cavernous Sinus) | Adults, F>M typically. Slow growing, dural-based (planum sphenoidale, tuberculum sellae, diaphragm, cavernous sinus wall). Mass effect (vision loss, CN palsies), headache, seizures. Rarely significant endocrine dysfunction unless very large. | Often isointense T1/T2, intense homogeneous enhancement after contrast, characteristic "dural tail" sign common. May cause hyperostosis (bone thickening) or encase vessels. |
| Rathke's Cleft Cyst | Benign epithelial cyst (remnant of Rathke's pouch), often incidental. May cause headache, visual disturbance, or hypopituitarism if large. Usually intrasellar or intra/suprasellar midline location. | Well-defined non-enhancing cyst (usually). Signal varies greatly (T1 hyper/iso/hypo, T2 usually hyper) based on proteinaceous content. Thin, smooth wall, usually no solid component or enhancement (may show thin rim). Intracystic nodule (proteinaceous/hemorrhagic) possible (<30%). |
| Pituitary Hyperplasia | Enlargement of normal gland tissue. Physiological (pregnancy, lactation, puberty) or pathological (response to end-organ failure like primary hypothyroidism, primary adrenal insufficiency). | Diffusely enlarged gland (>10-12mm height possible), usually enhances homogeneously and intensely. Sella may enlarge symmetrically. Gland shape maintained (concave superior border lost). Resolves if underlying cause treated. |
| Hypophysitis (Lymphocytic, Granulomatous, IgG4) | Inflammation of pituitary/stalk. Headache, visual disturbance, hypopituitarism (often rapid onset, multiple axes), Diabetes Insipidus (DI) common (esp. stalk involvement). Lymphocytic often postpartum/autoimmune context. | Often symmetrical gland/stalk enlargement and thickening, diffuse intense enhancement. May mimic adenoma but often more infiltrative appearance of stalk. Clinical context crucial (rapid onset hypopituitarism/DI). |
| Metastasis | History of primary cancer (breast, lung most common). Rapid onset hypopituitarism, DI common (posterior pituitary/stalk predilection). Headache, visual loss, CN palsies possible. | Enhancing lesion, often infiltrative, frequently involves posterior pituitary or stalk. May be multiple intracranial lesions. Search for primary malignancy is key. |
| Aneurysm (Cavernous/Suprasellar ICA) | Can mimic sellar mass, especially if partially thrombosed. May cause CN palsies (III, IV, V, VI due to cavernous sinus location), headache. Risk of rupture (SAH). | MRI shows characteristic flow void (if patent), laminated signal of thrombus (if present), enhancement of wall. MRA/CTA/DSA confirms diagnosis and delineates vascular anatomy. |
| Chordoma / Chondrosarcoma | Rare malignant/locally aggressive skull base tumors arising from notochord remnants (chordoma) or cartilage (chondrosarcoma). May extend into sella/suprasellar region from clivus/petrous apex. Often cause extensive bone destruction, CN palsies. | CT shows bone destruction. MRI shows locally destructive enhancing mass, often T2 hyperintense (esp. chordoma due to physaliphorous cells). Location centered on clivus or petroclival junction typically. |
| Germinoma (Suprasellar) | Rare germ cell tumor, usually children/young adults. Classic triad: DI, visual loss, hypopituitarism. May involve pineal region simultaneously ("dual midline tumors"). | Homogeneously enhancing suprasellar mass, often involves optic chiasm, stalk/hypothalamus. CSF/serum tumor markers (AFP, β-hCG) may be elevated (though often normal in pure germinoma). Highly radiosensitive. |
Diagnosis often involves Magnetic Resonance Imaging (MRI) of the pituitary gland, the gold standard for visualizing adenomas (microadenomas, macroadenomas, including prolactinomas), assessing their size, extent, relationship to surrounding structures like the optic chiasm and cavernous sinuses, and helping differentiate from other sellar lesions.
The diagnostic approach combines clinical assessment, hormonal testing, and imaging:
- Clinical Suspicion: Based on symptoms of hormone excess (e.g., acromegaly features, Cushingoid signs, galactorrhea/amenorrhea), mass effect (visual changes, headache, cranial nerve palsy), hypopituitarism (fatigue, hypogonadism, cold intolerance, etc.), or incidental finding on imaging.
- Hormonal Evaluation: Measurement of basal levels of anterior pituitary hormones and their target organ hormones is essential for identifying hormone excess or deficiency. Key tests include:
- Prolactin (PRL): Elevated levels suggest hyperprolactinemia. Markedly elevated levels (>200-250 ng/mL or mcg/L) are highly suggestive of a prolactinoma (especially macroadenoma). Moderately elevated levels (25-150 ng/mL) require evaluation for other causes (medications, hypothyroidism, stalk effect, etc.). Check for "hook effect" with diluted sample if macroadenoma present and PRL unexpectedly low/normal [24].
- Growth Hormone (GH) / Insulin-like Growth Factor 1 (IGF-1): Elevated age- and sex-matched IGF-1 is the best screening test for acromegaly. Diagnosis confirmed by failure of GH suppression (< 0.4 or < 1 ng/mL depending on assay) during an oral glucose tolerance test (OGTT). Basal GH levels are less reliable due to pulsatility.
- ACTH / Cortisol: Screening for Cushing's syndrome involves tests like late-night salivary cortisol, 24-hour urine free cortisol (UFC), or low-dose (1mg) overnight dexamethasone suppression test. If positive, plasma ACTH levels help differentiate ACTH-dependent (pituitary/ectopic) vs. independent (adrenal) causes. High-dose dexamethasone suppression tests and/or CRH stimulation tests, sometimes combined with inferior petrosal sinus sampling (IPSS), help confirm pituitary origin (Cushing's disease) [4].
- TSH / Free T4/T3: Elevated or inappropriately normal TSH with elevated free T4/T3 suggests a rare TSH-oma. Testing also screens for primary hypothyroidism (high TSH, low FT4), a potential cause of secondary hyperprolactinemia.
- LH / FSH / Testosterone (men) / Estradiol (women): Assess gonadal function. Low levels indicate hypogonadism (secondary if LH/FSH also low/normal). Elevated alpha-subunit common in gonadotroph adenomas.
- Assessment for Hypopituitarism: If mass effect or symptoms suggest deficiency, test baseline morning cortisol and ACTH, TSH and free T4, LH/FSH and testosterone/estradiol (as appropriate), IGF-1 (for GH axis). Dynamic stimulation tests (e.g., ACTH stimulation test, insulin tolerance test - gold standard but risky, GHRH-arginine test) may be needed to confirm subtle deficiencies. Assess for Diabetes Insipidus (DI) if posterior pituitary involved (polyuria, polydipsia, serum/urine osmolality).
- Imaging:
- Pituitary MRI: With and without gadolinium contrast, using dedicated thin slices (≤3mm) through the sella in coronal and sagittal planes. Dynamic contrast-enhanced sequences are particularly useful for identifying microadenomas, which often appear as focal areas of delayed enhancement compared to the briskly enhancing normal anterior pituitary gland. MRI precisely defines tumor size, relationship to optic chiasm, cavernous sinus invasion (Knosp classification), and stalk deviation [25].
- CT Scan: Less sensitive for adenomas themselves but better for showing bony erosion of the sella floor or clinoid processes, and calcifications (more common in craniopharyngiomas). Useful if MRI is contraindicated.
- Ophthalmological Evaluation: Formal visual field testing (typically automated perimetry, e.g., Humphrey visual field) is mandatory if an adenoma is near or compressing the optic chiasm (macroadenomas), even if the patient reports no visual symptoms. Assessment of visual acuity and optic nerve appearance (funduscopy for pallor/atrophy or papilledema) is also standard.
- Differential Diagnosis of Hyperprolactinemia: If elevated prolactin is found, systematically rule out other causes before diagnosing prolactinoma, especially if levels are only moderately high (<150 ng/mL) or imaging is normal/equivocal. Consider:
- Physiological causes: Pregnancy, lactation, stress, sleep (check morning levels), exercise, nipple stimulation, intercourse.
- Medications: Review current medications (antipsychotics - esp. risperidone, typicals; antidepressants - SSRIs, TCAs; antiemetics - metoclopramide, domperidone; antihypertensives - verapamil, methyldopa; opioids; estrogens). Stop suspect drug if possible and recheck PRL after ~3 days (or longer half-life).
- Systemic conditions: Primary hypothyroidism (check TSH/FT4), chronic kidney disease (check creatinine), severe liver cirrhosis, polycystic ovary syndrome (PCOS), chest wall lesions (herpes zoster, surgery scars, piercings).
- Pituitary stalk effect: Compression of the pituitary stalk by non-prolactin secreting tumors (NFPAs, craniopharyngiomas, meningiomas) or inflammatory lesions interrupts dopamine delivery from hypothalamus to lactotrophs, leading to mild/moderate hyperprolactinemia (usually <150 ng/mL). Imaging is key.
- Macroprolactinemia: Presence of large, biologically less active prolactin complexes; requires specific lab testing (PEG precipitation); clinically insignificant usually.
Normal serum prolactin levels vary slightly by lab and assay but generally are (approximate ranges):
Patient Group |
Approximate Normal Prolactin Range (ng/mL or mcg/L) |
(Approximate SI Units: mIU/L) |
|---|---|---|
Children (pre-pubertal) |
Variable, generally < 15-20 | ~ < 300-400 |
Adult Men |
< 15-20 | ~ < 300-400 |
Nonpregnant Adult Women |
< 25-30 | ~ < 500-600 |
Pregnant Women |
Significantly elevated, increases throughout pregnancy (e.g., up to 200-400+ ng/mL) | ~ up to 4000-8000+ mIU/L |
Postpartum (lactating) |
Elevated, levels fluctuate with breastfeeding, gradually decline if not nursing | Variable, can be very high initially |
Conversion factor is approximate (1 ng/mL ≈ 21 mIU/L based on WHO standards, but varies by assay).

Pituitary Adenoma and Hyperprolactinemia Syndrome Treatment
Treatment goals for pituitary adenomas are to normalize hormone levels (if excessive), relieve mass effect symptoms (visual loss, headache, cranial nerve palsies), preserve or restore normal pituitary function, and prevent tumor recurrence. The specific approach depends heavily on the adenoma type (functioning vs. nonfunctioning), size (micro vs. macro), hormone secreted, presence and severity of mass effect, patient age, reproductive goals, and comorbidities [26, 2, 4, 23].
- Prolactinomas (Hyperprolactinemia):
- Medical Therapy (First-line): Dopamine agonists (DAs) are highly effective and the preferred initial treatment for virtually all prolactinomas, including large macroadenomas causing visual field defects (unless vision loss is severe/rapidly progressive).
- Cabergoline: Generally preferred due to higher efficacy in normalizing prolactin and shrinking tumors, longer half-life allowing once or twice weekly dosing, and better tolerability compared to bromocriptine.
- Bromocriptine: Older agent, requires daily dosing, more associated with nausea/postural hypotension, but has a longer safety record, especially in pregnancy.
- Surgery (Neurosurgical intervention - Transsphenoidal Surgery): Indicated primarily for patients intolerant or resistant to dopamine agonists, those with pituitary apoplexy in a prolactinoma, rare cases with large cystic/hemorrhagic components unresponsive to DAs, or sometimes women desiring pregnancy who cannot tolerate or wish to avoid DAs. Cure rates are higher for microadenomas (~70-90%) than invasive macroadenomas (~30-50%).
- Radiotherapy (Stereotactic Radiosurgery or Fractionated RT): Reserved for rare cases of invasive/aggressive prolactinomas resistant or recurring after maximal medical and surgical therapy. Used primarily for tumor control, as effect on prolactin levels is slow and often incomplete; high risk of hypopituitarism.
- Medical Therapy (First-line): Dopamine agonists (DAs) are highly effective and the preferred initial treatment for virtually all prolactinomas, including large macroadenomas causing visual field defects (unless vision loss is severe/rapidly progressive).
- Acromegaly (GH-secreting adenomas):
- Surgery (Generally First-line): Transsphenoidal surgery is the primary treatment aiming for complete resection and biochemical cure (normalization of age/sex-matched IGF-1 and suppression of GH during OGTT). Cure rates depend on tumor size, invasiveness, and surgeon experience.
- Medical Therapy: Used for persistent disease after surgery, patients unsuitable for surgery, or sometimes preoperatively to improve surgical outcomes. Options include:
- Somatostatin Receptor Ligands (SRLs): First-generation (octreotide LAR, lanreotide Autogel) normalize IGF-1/GH in ~50-60% and may shrink tumors. Second-generation (pasireotide LAR) may be effective if first-gen SRLs fail but higher risk of hyperglycemia.
- GH Receptor Antagonist (Pegvisomant): Highly effective in normalizing IGF-1 (~80-90%) by blocking GH action at the receptor; does not shrink tumor and requires monitoring of liver function and tumor size.
- Dopamine Agonists (Cabergoline): Less effective, normalizes IGF-1 in only ~10-30%, mainly used as add-on therapy, especially if tumor co-secretes prolactin.
- Radiotherapy (Stereotactic or Fractionated): Effective for controlling tumor growth and slowly lowering GH/IGF-1 levels in patients with residual/recurrent disease unresponsive to medical therapy. High risk of delayed hypopituitarism.
- Cushing's Disease (ACTH-secreting adenomas):
- Surgery (First-line): Transsphenoidal selective adenomectomy is the treatment of choice, aiming for biochemical remission while preserving normal pituitary function. Remission rates are ~70-90% for microadenomas in experienced centers [27].
- Medical Therapy: Used preoperatively to control severe hypercortisolism, postoperatively for persistent/recurrent disease while awaiting effects of RT, or as primary therapy if surgery fails/contraindicated. Drugs target adrenal steroidogenesis (ketoconazole, metyrapone, etomidate, osilodrostat), pituitary ACTH secretion (pasireotide, cabergoline), or glucocorticoid receptor blockade (mifepristone).
- Radiotherapy (Stereotactic or Fractionated): Effective for persistent/recurrent disease after surgery, but biochemical remission is delayed (months to years).
- Bilateral Adrenalectomy: Provides definitive cure of hypercortisolism if pituitary-directed therapies fail, but results in permanent adrenal insufficiency requiring lifelong glucocorticoid/mineralocorticoid replacement and carries risk of Nelson's syndrome (progressive growth/pigmentation from the pituitary ACTH-secreting tumor).
- TSH-omas (rare):
- Surgery (First-line): Transsphenoidal resection is the preferred treatment.
- Medical Therapy: Somatostatin receptor ligands (octreotide, lanreotide) are often effective in suppressing TSH secretion and shrinking tumors preoperatively or for residual disease. Antithyroid drugs (methimazole, PTU) manage hyperthyroidism symptoms but don't treat the tumor.
- Radiotherapy: For residual/recurrent tumors unresponsive to other therapies.
- Nonfunctioning Pituitary Adenomas (NFPAs): See section below.
Management decisions should be individualized and often require a multidisciplinary team approach involving endocrinologists, neurosurgeons, neuroradiologists, ophthalmologists, and sometimes radiation oncologists.
Treatment of Nonfunctioning Pituitary Adenomas (NFPAs)
As NFPAs typically do not cause hormone excess syndromes, treatment is indicated primarily when they cause mass effect (visual impairment, cranial nerve palsies, clinically significant headaches attributable to the tumor) or hypopituitarism, or if they demonstrate significant growth on serial imaging [22, 23].
- Asymptomatic Incidental NFPAs:
- Microadenomas (<10mm): Generally managed conservatively with periodic MRI surveillance (e.g., MRI at 1 year, then 1-2 years, then less frequently if stable) and baseline endocrine evaluation (screening for hypopituitarism and hyperprolactinemia due to stalk effect). Most (~90-95%) do not grow significantly [8].
- Macroadenomas (≥10mm) without Mass Effect (No visual deficits, normal pituitary function, not abutting chiasm): Management is individualized. Options include close monitoring with MRI (e.g., at 6 months, then annually if stable) and regular endocrine/visual assessments, or consideration of surgery especially if the tumor is large or very close to the optic chiasm, given the potential for future growth/complications. Intervention is indicated if there is documented tumor growth, development of symptoms (visual loss, hypopituitarism), or if the tumor comes to abut/compress the optic chiasm [23].
- Symptomatic NFPAs (Causing Mass Effect or Hypopituitarism):
- Surgical Intervention (First-line): Transsphenoidal surgery is the primary treatment. The main goals are decompression of the optic apparatus to improve/preserve vision and maximal safe tumor removal to alleviate mass effect and potentially allow recovery of pituitary function (less likely if deficits are long-standing). Endoscopic endonasal approaches are now commonly used. Complete resection is often difficult for large or invasive (e.g., into cavernous sinus) tumors.
- Radiotherapy (Stereotactic Radiosurgery - SRS or Fractionated RT): Indicated primarily as an adjuvant treatment for significant residual tumor after surgery (especially if growing or large residual volume), or for recurrent tumors. Primary RT is rarely used unless surgery is contraindicated. RT provides excellent long-term tumor control rates (>90%) but does not immediately reduce mass effect and carries a significant delayed risk (~30-60% over 10 years) of radiation-induced hypopituitarism [28].
- Medical Therapy: There is currently no effective medical therapy proven to consistently shrink NFPAs or prevent growth. Dopamine agonists or somatostatin analogs have been studied but are generally ineffective for typical gonadotroph NFPAs.
- Management of Hypopituitarism: Hormone deficiencies identified before or after treatment require appropriate and lifelong physiological replacement therapy (e.g., hydrocortisone for adrenal insufficiency - stress dose coverage needed; levothyroxine for secondary hypothyroidism; testosterone/estrogen +/- progesterone for hypogonadism; potentially GH replacement in adults with confirmed deficiency and significant impact on quality of life or metabolic parameters; DDAVP for diabetes insipidus if present).
- Long-term Follow-up: Essential after treatment (surgery +/- radiation) for all macroadenomas. Includes monitoring for tumor recurrence or regrowth (serial MRI, frequency depends on residual tumor/treatment type), regular assessment of endocrine function (annual testing for hypopituitarism, including after RT), and monitoring visual status (visual fields if history of compression). Recurrence rates after surgery alone can be significant over time (10-50% depending on follow-up duration and residual tumor), necessitating consideration of adjuvant radiotherapy for concerning residual tumor.
![]() Attention! Pituitary adenomas require specialized diagnosis and management by a multidisciplinary team. Symptoms like vision changes, persistent headaches, hormonal imbalances (menstrual changes, lactation, unexplained physical changes, sexual dysfunction), or excessive fatigue warrant evaluation by a physician, potentially leading to referral to an endocrinologist and neurosurgeon.
Attention! Pituitary adenomas require specialized diagnosis and management by a multidisciplinary team. Symptoms like vision changes, persistent headaches, hormonal imbalances (menstrual changes, lactation, unexplained physical changes, sexual dysfunction), or excessive fatigue warrant evaluation by a physician, potentially leading to referral to an endocrinologist and neurosurgeon.
References
- Melmed S. Pituitary-Tumor Endocrinopathies. N Engl J Med. 2020 Mar 5;382(10):937-950. doi: 10.1056/NEJMra1810772
- Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011 Feb;96(2):273-88. doi: 10.1210/jc.2010-1692
- Katznelson L, Laws ER Jr, Melmed S, et al; Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Nov;99(11):3933-51. doi: 10.1210/jc.2014-2700
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008 May;93(5):1526-40. doi: 10.1210/jc.2008-0125
- Beck-Peccoz P, Persani L. Thyrotropin-secreting pituitary adenomas. N Engl J Med. 1996;334(11):712-713. doi: 10.1056/NEJM199603143341107
- Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2011 Jun;14(2):111-8. doi: 10.1007/s11102-011-0300-1
- Molitch ME. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 1997 Mar;26(1):1-17. doi: 10.1016/s0889-8529(05)70239-7
- Fernández-Balsells MM, Murad MH, Barwise A, et al. Clinical management of nonfunctioning pituitary adenomas (NFPAs): a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011 Apr;96(4):905-15. doi: 10.1210/jc.2010-1921
- Chapter on Pituitary Imaging. In: Osborn AG, Salzman KL, Jhaveri MD, et al. Osborn's Brain. 2nd ed. Elsevier; 2018.
- Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. 2002 Feb;2(2):83-94. doi: 10.1038/nrc760
- Vierimaa O, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006 May 26;312(5777):1228-30. doi: 10.1126/science.1126100
- Reincke M, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. 2015 Jan;47(1):31-8. doi: 10.1038/ng.3166
- Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936 Mar;12(2):205-16.1.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004 Aug 1;101(3):613-9. doi: 10.1002/cncr.20412
- Hall WA, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994 May 15;120(10):817-20. doi: 10.7326/0003-4819-120-10-199405150-00001
- Ciccarelli A, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Varese, Italy. J Clin Endocrinol Metab. 2001 Feb;86(2):720-5. doi: 10.1210/jcem.86.2.7204
- Fernandez A, et al. Prevalence of pituitary adenomas: a community-based study in Olmsted County, Minnesota, 1993-1997. Arch Intern Med. 2000 Jan 10;160(1):78-81. doi: 10.1001/archinte.160.1.78
- Daly AF, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006 Dec;91(12):4756-69. doi: 10.1210/jc.2006-1668
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009 Mar;119(3):429-38. doi: 10.1172/JCI38090
- Nieman LK. Cushing's syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015 Oct;173(4):M33-8. doi: 10.1530/EJE-15-0464
- Elster AD, et al. Pituitary gland: MR imaging of physiologic hypertrophy in adolescence. Radiology. 1990;174(3 Pt 1):681-5. doi: 10.1148/radiology.174.3.2305044
- Molitch ME. Nonfunctioning pituitary adenomas. Pituitary. 2005;8(3-4):213-20. doi: 10.1007/s11102-006-8512-6
- Freda PU, Beckers AM, Katznelson L, et al; Endocrine Society. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011 Apr;96(4):894-904. doi: 10.1210/jc.2010-1048
- Frieze TW, et al. Hook effect in prolactinomas: case report and review of literature. Endocr Pract. 2006;12(3):296-303. doi: 10.4158/EP.12.3.296
- Bonneville F, et al. Radiology of pituitary adenomas: an update. Radiol Clin North Am. 2012 Nov;50(6):995-1013. doi: 10.1016/j.rcl.2012.08.005
- Melmed S, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509-17. doi: 10.1210/jc.2008-2421
- Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818
- Sheehan JP, et al. Radiosurgery for nonfunctioning pituitary adenomas: a systematic review. J Neurosurg. 2013 Aug;119(2):446-56. doi: 10.3171/2013.4.JNS121833
See also
- Anatomy of the nervous system
- Central nervous system infection:
- Brain abscess (lobar, cerebellar)
- Eosinophilic granuloma, Langerhans cell histiocytosis (LCH), Hennebert's symptom
- Epidural brain abscess
- Sinusitis-associated intracranial complications
- Otogenic intracranial complications
- Sinusitis-associated ophthalmic complications
- Bacterial otogenic meningitis
- Subdural brain abscess
- Sigmoid sinus suppurative thrombophlebitis
- Cerebral 3rd Ventricle Colloid Cyst
- Cerebral and spinal adhesive arachnoiditis
- Corticobasal Ganglionic Degeneration (Limited Brain Atrophy)
- Encephalopathy
- Headache, migraine
- Traumatic brain injury (concussion, contusion, brain hemorrhage, axonal shearing lesions)
- Increased intracranial pressure and hydrocephalus
- Parkinson's disease
- Pituitary microadenoma, macroadenoma and nonfunctioning adenomas (NFPAs), hyperprolactinemia syndrome
- Spontaneous cranial cerebrospinal fluid leak (CSF liquorrhea)