Microadenoma hipofisario, macroadenoma y adenomas no funcionantes (NFPAs), síndrome de hiperprolactinemia
- Resumen del adenoma hipofisario (microadenoma, macroadenoma) y síndrome de hiperprolactinemia
- Microadenomas Hipofisarios:
- Macroadenomas Hipofisarios:
- Adenomas Hipofisarios No Funcionantes (NFPAs)
- Diagnóstico del Adenoma Hipofisario y Síndrome de Hiperprolactinemia
- Tratamiento del Adenoma Hipofisario y Síndrome de Hiperprolactinemia
- Tratamiento de los Adenomas Hipofisarios No Funcionantes (NFPAs)
- Referencias
Resumen del adenoma hipofisario (microadenoma, macroadenoma) y síndrome de hiperprolactinemia
Los adenomas hipofisarios son típicamente tumores benignos (no cancerosos) que se originan en las células productoras de hormonas de la glándula pituitaria anterior (adenohipófisis), ubicada en la base del cerebro dentro de una estructura ósea llamada silla turca [1]. Estos adenomas se clasifican según su tamaño (microadenoma frente a macroadenoma) y su actividad hormonal (funcionantes frente a no funcionantes).
El síndrome de hiperprolactinemia se refiere a las consecuencias clínicas de los niveles persistentemente elevados de prolactina, una hormona normalmente implicada en la lactancia. Los prolactinomas (adenomas hipofisarios secretores de prolactina) son el tipo más común de adenoma hipofisario funcionante y una causa frecuente de hiperprolactinemia [2]. En las mujeres, los síntomas suelen incluir galactorrea (producción inapropiada de leche), irregularidades menstruales (oligomenorrea o amenorrea) e infertilidad. En los hombres, la hiperprolactinemia a menudo conduce a la disminución de la libido, disfunción eréctil, infertilidad debida a oligospermia, ginecomastia (agrandamiento de los senos) y, con menor frecuencia, galactorrea. La prolactina elevada inhibe la secreción de la hormona liberadora de gonadotropina (GnRH) desde el hipotálamo, lo que lleva a un hipogonadismo secundario.

Los síntomas relacionados con los adenomas hipofisarios surgen ya sea por un exceso en la secreción de hormonas (en los adenomas funcionantes) o por el tamaño físico del tumor y la presión sobre las estructuras circundantes (efecto de masa), especialmente en el caso de los macroadenomas. Los síndromes de exceso hormonal incluyen:
Hormona en Exceso |
Condición Resultante y Principales Problemas de Salud |
|---|---|
| Prolactina (PRL) |
Hiperprolactinemia: - Mujeres: Disfunción menstrual (oligomenorrea, amenorrea), galactorrea, infertilidad, disminución de la densidad ósea. - Hombres: Disminución de la libido, disfunción eréctil, infertilidad, ginecomastia (menos común), disminución de la densidad ósea. [2] |
| Hormona del Crecimiento (GH) |
Acromegalia (adultos) / Gigantismo (niños): - Agrandamiento de manos, pies y rasgos faciales (mandíbula, frente). - Hinchazón de tejidos blandos, sudoración excesiva. - Osteoartritis, síndrome del túnel carpiano. - Apnea del sueño. - Hipertensión, miocardiopatía. - Diabetes mellitus o intolerancia a la glucosa. - Mayor riesgo de pólipos/cáncer colorrectal. [3] |
| Hormona Adrenocorticotrópica (ACTH) |
Enfermedad de Cushing (Síndrome de Cushing dependiente de la hipófisis): - Obesidad central (grasa troncal, cara de luna llena, joroba de búfalo), piel fina, facilidad para los hematomas, estrías púrpuras. - Debilidad muscular (miopatía proximal), osteoporosis. - Cambios de humor (ansiedad, depresión, psicosis). - Hipertensión, diabetes mellitus. - Mayor susceptibilidad a infecciones. - Irregularidades menstruales (mujeres). [4] |
| Hormona Estimulante de la Tiroides (TSH) |
TSH-oma (raro): - Hipertiroidismo Secundario: Los síntomas incluyen intolerancia al calor, sudoración, taquicardia/palpitaciones (arritmias como fibrilación auricular), temblores, ansiedad/irritabilidad, pérdida de peso involuntaria a pesar de un mayor apetito, bocio. Caracterizado por TSH inapropiadamente normal o elevada con niveles altos de T4/T3 libre. [5] |
| Gonadotropinas (LH, FSH) |
Adenoma Gonadotropo (a menudo clínicamente no funcionante): - Generalmente secretan fragmentos de hormonas inactivos o intactos (p. ej., subunidad alfa, beta-FSH, con menor frecuencia FSH/LH intacta). - Rara vez pueden causar hiperestimulación ovárica (mujeres) o agrandamiento testicular/pubertad precoz (hombres). - A menudo se presentan debido a un efecto de masa o se descubren de forma incidental. [6] |
Los adenomas hipofisarios no funcionantes (NFPAs) no secretan cantidades clínicamente significativas de hormonas activas, aunque muchos producen subunidades hormonales detectables mediante pruebas especiales. Por lo general, causan síntomas debido al efecto de masa cuando se convierten en macroadenomas, comprimiendo las estructuras adyacentes como el quiasma óptico (causando defectos del campo visual, clásicamente hemianopsia bitemporal), los nervios craneales dentro del seno cavernoso (causando visión doble) o el tejido pituitario normal (causando hipopituitarismo, que es la deficiencia de una o más hormonas pituitarias). Los dolores de cabeza también son comunes debido al estiramiento de la duramadre o al aumento de la presión selar.
Microadenomas Hipofisarios
Descripción de los Microadenomas Hipofisarios
Un microadenoma hipofisario se define como un adenoma de la glándula pituitaria con un diámetro máximo de menos de 10 milímetros (menos de 1 cm) [7]. Estos tumores están confinados dentro de la silla turca y típicamente no causan síntomas de efecto de masa como pérdida de visión. Los microadenomas pueden ser funcionantes (secretores de hormonas) o no funcionantes. Los microadenomas funcionantes provocan síntomas relacionados con la hormona específica que sobreproducen (por ejemplo, el prolactinoma causa hiperprolactinemia, el secretor de ACTH causa la enfermedad de Cushing, y el secretor de GH causa acromegalia).
Muchos microadenomas, sin embargo, son clínicamente no funcionantes y se descubren de manera incidental durante pruebas de imagen cerebral (Imágenes por Resonancia Magnética - RM o TC) realizadas por otras razones (p. ej., dolor de cabeza, mareos, trauma). A menudo se les conoce como 'incidentalomas' hipofisarios. Si bien la gran mayoría son hallazgos benignos que solo requieren seguimiento, incluso los pequeños microadenomas incidentales a veces pueden causar desequilibrios hormonales sutiles (Cushing subclínico, hiperprolactinemia leve) o, rara vez (< 10 % en varios años), crecer con el tiempo [8].
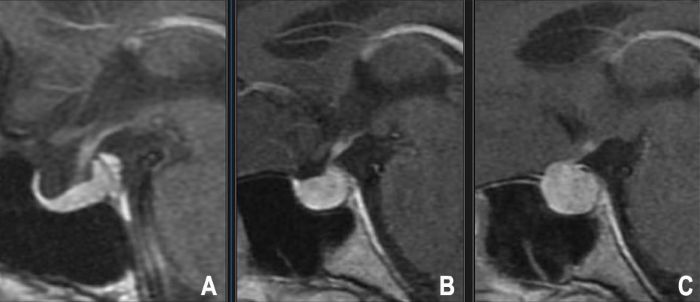
La RM con contraste de gadolinio, particularmente utilizando secuencias dinámicas realzadas con contraste y cortes finos a través de la fosa pituitaria, es la modalidad de imagen preferida para detectar microadenomas. A menudo aparecen como lesiones focales pequeñas que se realzan menos o más tarde que el tejido normal circundante de la glándula pituitaria (típicamente hipointensas en las imágenes tempranas T1 poscontraste) [9].
Los microadenomas clínicamente significativos son más comúnmente prolactinomas, seguidos de los adenomas secretores de ACTH que causan la enfermedad de Cushing, y los adenomas secretores de GH que causan acromegalia. Los microadenomas no funcionantes a menudo son adenomas gonadotropos por inmunohistoquímica, pero carecen de exceso hormonal clínico. El diagnóstico diferencial en imágenes incluye quistes de la hendidura de Rathke, hiperplasia pituitaria, metástasis o procesos inflamatorios (hipofisitis), aunque estos a menudo presentan características o contextos clínicos distintos.
Fisiopatología de los Microadenomas Hipofisarios
Los adenomas hipofisarios, incluyendo los microadenomas, se consideran generalmente neoplasias monoclonales, lo que significa que surgen de la expansión clonal de una única célula de la adenohipófisis mutada [10]. Las mutaciones iniciales específicas y los eventos genéticos/epigenéticos subsecuentes que conducen a la formación del adenoma son complejos y variados, pero están implicados múltiples factores y vías.
Existen síndromes genéticos germinales específicos que predisponen a los individuos a los adenomas hipofisarios: la Neoplasia Endocrina Múltiple tipo 1 (MEN1), causada por mutaciones en el gen supresor de tumores MEN1, frecuentemente involucra prolactinomas o adenomas secretores de GH. El síndrome de McCune-Albright, debido a mutaciones activadoras en mosaico en el gen GNAS que codifica la subunidad de la proteína Gs-alfa, a menudo conduce a adenomas secretores de GH (somatotropinomas). El complejo de Carney (mutaciones PRKAR1A) puede incluir adenomas pituitarios (típicamente secretores de GH). Las mutaciones en el gen AIP predisponen a adenomas hipofisarios aislados familiares (FIPA), particularmente tumores secretores de GH o prolactina, que a menudo se presentan a una edad temprana y son más grandes/agresivos [11].
Los adenomas esporádicos (no familiares) también involucran alteraciones genéticas y epigenéticas somáticas. Las mutaciones somáticas activadoras en GNAS se encuentran en aproximadamente el 40 por ciento de los adenomas secretores de GH esporádicos. Las mutaciones en USP8 son comunes en los adenomas secretores de ACTH [12]. Otras mutaciones somáticas y cambios epigenéticos que afectan los reguladores del ciclo celular (p. ej., pérdida de función en CDKN1B/p27, alteraciones en p53, vías RB), vías de factores de crecimiento (p. ej., sobreexpresión de FGFR4) y factores de transcripción específicos de la hipófisis contribuyen a la tumorigénesis y la producción hormonal. También se observa pérdida de heterocigosidad en ciertas ubicaciones cromosómicas (p. ej., 11q13 donde reside MEN1).
Los prolactinomas, el tipo más común, surgen de las células lactotropas. Los corticotropinomas que causan la enfermedad de Cushing surgen de las células corticotropas. Los somatotropinomas que causan acromegalia surgen de las células somatotropas. Los gonadotropinomas suelen surgir de las células gonadotropas y a menudo secretan subunidades hormonales inactivas (subunidad alfa, beta-FSH, beta-LH). Los tirotropinomas que causan hipertiroidismo surgen de las células tirotropas y son raros.
La mayoría de los adenomas clínicamente no funcionantes son inmunopositivos para las subunidades de gonadotropina (FSH, LH, subunidad alfa) pero no causan síndromes clínicos de exceso hormonal. Algunos pueden ser adenomas de "células nulas" sin tinción hormonal específica, o adenomas corticotropos/somatotropos silenciosos.
Epidemiología de los Microadenomas Hipofisarios
Frecuencia de los Microadenomas Hipofisarios
Los microadenomas hipofisarios son hallazgos incidentales sorprendentemente comunes. Los estudios de autopsias han reportado adenomas microscópicos o pequeños (a menudo < 3 mm) en hasta el 27 por ciento de los individuos sin enfermedad hipofisaria conocida [13, 14]. Los estudios de imágenes de alta resolución (RM) detectan incidentalomas pituitarios (la mayoría de los cuales son microadenomas < 10 mm) en aproximadamente el 10-20 por ciento de la población general que se somete a exploraciones cerebrales por razones no relacionadas [15].
La gran mayoría de estos microadenomas descubiertos incidentalmente son no funcionantes y se mantienen estables o crecen muy lentamente, sin causar nunca síntomas clínicos. Los microadenomas funcionantes son mucho menos comunes, pero son la causa habitual de síndromes de exceso de hormonas pituitarias como la hiperprolactinemia (siendo el prolactinoma el tipo diagnosticado con mayor frecuencia, con una prevalencia estimada de ~10-50 por 100,000 habitantes [16]), la enfermedad de Cushing y la acromegalia.
Los microadenomas hipofisarios pueden ocurrir a cualquier edad, pero se diagnostican con mayor frecuencia en adultos jóvenes y de mediana edad (20s-50s). Hay un predominio femenino para los prolactinomas (proporción M:H ~10:1) y la enfermedad de Cushing (~3-5:1), mientras que los adenomas secretores de GH tienen una distribución de género aproximadamente equitativa.
Antes del uso generalizado de la RM, la hiperprolactinemia detectada mediante análisis de sangre era una pista importante para descubrir microadenomas secretores de prolactina subyacentes, a menudo invisibles. La RM ahora permite la visualización directa.
La alta prevalencia de microadenomas hallados incidentalmente en comparación con la menor prevalencia de enfermedad hipofisaria clínicamente significativa o macroadenomas sugiere que la mayoría de los microadenomas no progresan hasta convertirse en tumores grandes o sintomáticos.
Macroadenomas Hipofisarios
Descripción de los Macroadenomas Hipofisarios
Un macroadenoma hipofisario se define como un adenoma pituitario que mide 10 milímetros (1 cm) o más en su diámetro máximo [3]. A diferencia de los microadenomas, los macroadenomas frecuentemente se extienden más allá de los límites de la silla turca, pudiendo comprimir las estructuras neurales y vasculares circundantes.
Pueden ser funcionantes (secretores de hormonas) o no funcionantes. Los adenomas hipofisarios no funcionantes (NFPAs) son el tipo más común de macroadenoma diagnosticado clínicamente [6]. Los macroadenomas funcionantes incluyen prolactinomas grandes, adenomas secretores de GH que causan acromegalia y, con menor frecuencia, adenomas grandes secretores de ACTH (a menudo asociados con el síndrome de Nelson después de la adrenalectomía) o adenomas secretores de TSH.
Los macroadenomas a menudo se presentan con síntomas relacionados con el efecto de masa debido a su tamaño:
- Alteraciones visuales: La compresión del quiasma óptico, situado en la parte superior de la hipófisis, es común cuando existe extensión supraselar, lo que clásicamente causa hemianopsia bitemporal (pérdida de visión periférica en ambos campos temporales exteriores). La compresión de los nervios ópticos o tractos puede causar otros patrones de pérdida de visión o agudeza visual reducida.
- Parálisis de los nervios craneales: La extensión lateral hacia el seno cavernoso puede comprimir los nervios craneales III (oculomotor), IV (troclear), V (trigémino - ramas oftálmica V1 y maxilar V2) y VI (abducens), lo que provoca visión doble (diplopía), caída del párpado (ptosis), cambios pupilares o entumecimiento/dolor facial. Los NC III y VI son los más comúnmente afectados clínicamente.
- Dolores de cabeza: A menudo inespecíficos, posiblemente debido al estiramiento de la duramadre que cubre la silla turca (diaphragma sellae) o al aumento de la presión intraselar.
- Hipopituitarismo: La compresión y destrucción del tejido pituitario normal puede conducir a una deficiencia de una o más hormonas de la hipófisis anterior (la deficiencia de GH suele ser la más temprana, seguida de las gonadotropinas LH/FSH que causan hipogonadismo, luego la TSH que causa hipotiroidismo secundario, y finalmente la ACTH que causa insuficiencia suprarrenal secundaria) o, menos comúnmente, disfunción de la hipófisis posterior (deficiencia de la hormona antidiurética - ADH, que causa diabetes insípida central, vista con mayor frecuencia en la compresión del tallo o lesiones originadas posteriormente).
- Apoplejía hipofisaria: Una emergencia médica aguda causada por una hemorragia o infarto repentino dentro de un adenoma hipofisario (mucho más común en los macroadenomas que en los microadenomas). Se presenta con dolor de cabeza intenso y repentino, pérdida rápida de la visión, parálisis de los nervios craneales, alteración del estado de conciencia y, a menudo, hipopituitarismo agudo (que requiere terapia de reemplazo con esteroides de manera urgente).
Los macroadenomas funcionantes también provocan síntomas relacionados con la sobreproducción de hormonas, a menudo más graves que en los microadenomas debido a la mayor carga tumoral, tal como se describió anteriormente.
Fisiopatología de los Macroadenomas Hipofisarios
La fisiopatología subyacente implica la expansión clonal de células mutadas de la adenohipófisis, similar a los microadenomas. Los factores que impulsan la progresión de microadenoma a macroadenoma o el desarrollo de novo de macroadenomas no se han elucidado por completo, pero probablemente involucran la acumulación de alteraciones genéticas adicionales (p. ej., mutaciones en genes supresores de tumores como p53, inestabilidad cromosómica) o epigenéticas que promueven un aumento en la proliferación, disminución de la apoptosis, angiogénesis y, en ocasiones, invasividad [10].
Los macroadenomas a menudo demuestran pérdida de los mecanismos normales de retroalimentación que controlan la secreción de hormonas (especialmente en los funcionantes). Su crecimiento conduce a la compresión y destrucción de las células pituitarias normales adyacentes, lo que resulta en hipopituitarismo. La extensión superior comprime el quiasma óptico y potencialmente el hipotálamo (lo que lleva a DI o desregulación del apetito/temperatura). La extensión lateral invade el seno cavernoso, potencialmente envolviendo la arteria carótida interna y los nervios craneales III, IV, V1, V2 y VI. La extensión inferior puede erosionar el piso de la silla turca hacia el seno esfenoidal, causando a veces rinorrea de líquido cefalorraquídeo (LCR).
La base genética involucra factores mencionados para los microadenomas, incluidos los síndromes germinales (MEN1, GNAS, AIP, complejo de Carney) y las mutaciones somáticas (GNAS, USP8). Otras alteraciones asociadas con tumores más grandes o más agresivos podrían incluir mutaciones en los reguladores del ciclo celular (p53, RB), receptores del factor de crecimiento (FGFR4) o moléculas de adhesión. Aunque histológicamente son benignos (baja tasa mitótica, generalmente sin atipia citológica), algunos macroadenomas exhiben patrones de crecimiento invasivos hacia las estructuras circundantes (duramadre, hueso, seno cavernoso), lo que dificulta la resección quirúrgica completa. Los verdaderos carcinomas pituitarios primarios, definidos por la presencia de metástasis craneoespinales o sistémicas, son extremadamente raros.
Síndromes clínicos asociados como MEN1, McCune-Albright y el complejo de Carney pueden implicar macroadenomas, a menudo del tipo secretor de GH o prolactina.
Epidemiología de los Macroadenomas Hipofisarios
Frecuencia de los Macroadenomas Hipofisarios
Si bien los microadenomas incidentales son muy comunes, los tumores pituitarios clínicamente significativos que requieren evaluación y tratamiento potencial son menos frecuentes. La prevalencia general de los adenomas hipofisarios clínicamente aparentes se estima en alrededor de 78-94 por cada 100,000 individuos, o aproximadamente 1 de cada 1000 [17, 18]. Los macroadenomas constituyen una proporción significativa de estos tumores clínicamente relevantes, representando probablemente el 30-50% de los adenomas diagnosticados, particularmente entre los adenomas no funcionantes que a menudo se presentan debido a su tamaño.
Los estudios poblacionales sugieren una incidencia anual global de adenomas hipofisarios recién diagnosticados de alrededor de 4 por 100,000 habitantes [18], con tasas de prevalencia en aumento debido a una mejor detección gracias a las imágenes. La incidencia que requiere una intervención neuroquirúrgica varía de aproximadamente 1 a 7 por 100,000 por año, siendo los macroadenomas que causan efecto de masa o exceso de hormonas refractario al tratamiento médico las indicaciones primarias para la cirugía.
Morbilidad y Mortalidad en Macroadenomas Hipofisarios
La morbilidad asociada con los macroadenomas hipofisarios proviene principalmente de los efectos de masa (pérdida visual, parálisis de los nervios craneales, dolores de cabeza debilitantes), disfunción hormonal (hipopituitarismo que requiere reemplazo de por vida, o las consecuencias sistémicas de los síndromes de exceso hormonal como la acromegalia o la enfermedad de Cushing) y las posibles complicaciones del tratamiento (p. ej., riesgos quirúrgicos como fuga de LCR, meningitis, lesión vascular, empeoramiento del hipopituitarismo; efectos a largo plazo de la radiación como hipopituitarismo, tumores secundarios, cambios cognitivos). La discapacidad visual, en particular los déficits permanentes derivados del tratamiento tardío de la compresión quiasmática, afecta significativamente la calidad de vida. El hipopituitarismo requiere un manejo cuidadoso y conlleva sus propios riesgos si no se trata de forma adecuada (p. ej., crisis suprarrenal). Las complicaciones del exceso hormonal incontrolado (p. ej., enfermedad cardiovascular, diabetes, osteoporosis, mayor riesgo de cáncer en acromegalia y Cushing) contribuyen de manera importante a la morbilidad y mortalidad a largo plazo si no se manejan eficazmente [19, 20].
La mortalidad directamente relacionada con la naturaleza benigna de los macroadenomas hipofisarios en sí es generalmente baja. Sin embargo, está bien documentado un mayor riesgo de mortalidad en pacientes con acromegalia no controlada y enfermedad de Cushing, principalmente debido a complicaciones cardiovasculares (hipertensión, insuficiencia cardíaca, accidente cerebrovascular, arritmias), problemas metabólicos (diabetes) e infecciones (Cushing). La apoplejía hipofisaria, aunque rara, puede ser fatal si no se reconoce y se trata a tiempo. Las complicaciones de la cirugía (aunque la mortalidad es baja, <1%, en centros experimentados) o los efectos a largo plazo de la radiación también pueden contribuir. La supervivencia general de los pacientes con macroadenomas es buena por lo general con un manejo multidisciplinario moderno que controla los niveles hormonales y el efecto de masa, aunque la calidad de vida puede verse afectada por síntomas residuales o efectos secundarios del tratamiento.
Los macroadenomas hipofisarios no muestran predilección racial significativa.
Si bien los estudios de autopsias muestran una prevalencia igual de adenomas incidentales entre sexos, los prolactinomas diagnosticados clínicamente y los adenomas secretores de ACTH (enfermedad de Cushing) son significativamente más comunes en mujeres en edad reproductiva, posiblemente debido a un reconocimiento más temprano de los síntomas (irregularidades menstruales, galactorrea). Los adenomas secretores de GH (acromegalia) y los NFPA que se presentan clínicamente tienen una distribución por sexos más equitativa. Los macroadenomas hipofisarios pueden aparecer a cualquier edad, pero se diagnostican con mayor frecuencia en adultos, con una incidencia máxima entre la tercera y sexta décadas.
Adenomas Hipofisarios No Funcionantes (NFPAs)
Los adenomas hipofisarios no funcionantes (NFPAs) son tumores de la hipófisis que no causan un síndrome clínico de hipersecreción hormonal. Representan aproximadamente el 15-30 por ciento de todos los adenomas hipofisarios diagnosticados clínicamente y son el tipo más común que se presenta como macroadenomas [22]. Aunque se denominan "no funcionantes", la tinción inmunohistoquímica revela que la mayoría de los NFPAs se originan a partir de células gonadotropas y con frecuencia producen hormonas gonadotropinas (Hormona Foliculoestimulante - FSH, Hormona Luteinizante - LH) o sus subunidades (especialmente la subunidad alfa o la subunidad beta de la FSH). Sin embargo, estas hormonas suelen ser biológicamente inactivas, se secretan de manera ineficiente o en proporciones anormales, por lo que no causan síntomas clínicos de exceso hormonal [6]. Algunos pueden ser verdaderos adenomas de "células nulas" o variantes silentes de otras células productoras de hormonas.
Debido a que carecen de síndromes de hipersecreción hormonal, los NFPAs suelen presentarse más tarde que los adenomas funcionantes, a menudo cuando han crecido lo suficiente (macroadenomas) para causar síntomas debidos al efecto de masa:
- Defectos del campo visual (típicamente hemianopsia bitemporal por compresión del quiasma óptico).
- Dolores de cabeza (inespecíficos, debidos al estiramiento de la duramadre o la presión en la silla).
- Hipopituitarismo (por compresión del tejido pituitario normal, afectando con mayor frecuencia a la secreción de GH y gonadotropinas primero, seguidas por TSH y ACTH).
- Parálisis de pares craneales (menos común, por invasión del seno cavernoso, causando diplopía o entumecimiento facial).
- Apoplejía hipofisaria (hemorragia/infarto agudo dentro del tumor).
Muchos NFPAs más pequeños se descubren de forma incidental (RM o TC) como incidentalomas hipofisarios. Los estudios de autopsia y de imagen sugieren una alta prevalencia de microadenomas silenciosos (hasta 1 de cada 6 personas puede albergar uno), pero la mayoría permanece pequeños y asintomáticos durante toda la vida.
El tratamiento primario para los NFPAs sintomáticos que causan efecto de masa o hipopituitarismo progresivo es la resección quirúrgica, habitualmente a través de la ruta transesfenoidal. Los principales objetivos son la descompresión de las estructuras circundantes (especialmente el aparato óptico para mejorar/preservar la visión) y la eliminación máxima segura del tumor para reducir el efecto de masa y potencialmente mejorar la función pituitaria. El manejo de los NFPAs incidentales asintomáticos consiste en el seguimiento con imágenes seriadas y evaluaciones endocrinas/visuales [23].
Diagnóstico del Adenoma Hipofisario y Síndrome de Hiperprolactinemia
Diagnóstico Diferencial de Masas Selares/Supraselares
| Condición | Características Clave / Puntos Distintivos | Hallazgos Típicos en Imágenes (RM) |
|---|---|---|
| Adenoma Hipofisario (Micro/Macro) | Síntomas endocrinos (exceso/deficiencia) o efecto de masa (pérdida de visión, dolor de cabeza, parálisis de NC). Se origina dentro de la silla turca, desplaza la glándula, puede extenderse supra/paraselar. | Micro: Lesión focal <10mm, típicamente hipointensa en T1, se realza menos/más tarde que la glándula normal. Macro: Masa selar ≥10mm, realce variable (a menudo menor que la glándula), puede mostrar hemorragia/quiste, se extiende supra/paraselar, puede erosionar hueso. |
| Craneofaringioma | Pico de edad bimodal (niños/adultos mayores). Ubicación supraselar común. Dolores de cabeza, pérdida visual, disfunción endocrina (def de GH, DI común), hidrocefalia. | A menudo componentes mixtos quísticos (T1 variable/T2 hiper) y sólidos. Calcificación muy común (TC es mejor). Se realza (partes sólidas, pared del quiste gruesa). Típicamente epicentro supraselar, puede adherirse a estructuras circundantes. |
| Meningioma (Selar/Supraselar/Seno Cavernoso) | Adultos, M>H típicamente. Crecimiento lento, base dural (planum sphenoidale, tuberculum sellae, diafragma, pared del seno cavernoso). Efecto de masa (pérdida de visión, parálisis de NC), dolores de cabeza, convulsiones. Rara vez disfunción endocrina significativa a menos que sea muy grande. | A menudo isointenso en T1/T2, realce intenso y homogéneo tras el contraste, signo de "cola dural" característico común. Puede causar hiperostosis (engrosamiento óseo) o englobar vasos. |
| Quiste de la Hendidura de Rathke | Quiste epitelial benigno (remanente de la bolsa de Rathke), a menudo incidental. Puede causar dolor de cabeza, alteración visual o hipopituitarismo si es grande. Ubicación generalmente intraselar o intra/supraselar en la línea media. | Quiste bien definido que no se realza (generalmente). La señal varía enormemente (T1 hiper/iso/hipo, T2 usualmente hiper) según el contenido proteínico. Pared delgada y lisa, usualmente sin componente sólido o realce (puede mostrar anillo fino). Nódulo intraquístico (proteínico/hemorrágico) posible (<30%). |
| Hiperplasia Hipofisaria | Agrandamiento del tejido normal de la glándula. Fisiológica (embarazo, lactancia, pubertad) o patológica (respuesta a fallo de órgano terminal como hipotiroidismo primario, insuficiencia suprarrenal primaria). | Glándula agrandada de forma difusa (puede superar los 10-12 mm de altura), generalmente se realza de manera homogénea e intensa. La silla turca puede agrandarse simétricamente. Se mantiene la forma de la glándula (se pierde el borde superior cóncavo). Se resuelve si se trata la causa subyacente. |
| Hipofisitis (Linfocítica, Granulomatosa, IgG4) | Inflamación de la hipófisis/tallo. Dolor de cabeza, alteración visual, hipopituitarismo (a menudo de inicio rápido, múltiples ejes), la Diabetes Insípida (DI) es común (especialmente con afectación del tallo). La linfocítica se da a menudo en un contexto posparto/autoinmune. | A menudo agrandamiento y engrosamiento simétrico de la glándula/tallo, realce intenso y difuso. Puede imitar un adenoma, pero a menudo tiene una apariencia más infiltrativa en el tallo. El contexto clínico es crucial (hipopituitarismo de inicio rápido/DI). |
| Metástasis | Historia de cáncer primario (mama y pulmón son los más comunes). Hipopituitarismo de inicio rápido, DI es común (predilección por hipófisis posterior/tallo). Dolor de cabeza, pérdida de visión, parálisis de NC posibles. | Lesión que capta contraste, a menudo infiltrativa, frecuentemente afecta la hipófisis posterior o el tallo. Pueden ser múltiples lesiones intracraneales. La búsqueda de la malignidad primaria es clave. |
| Aneurisma (ACI Cavernosa/Supraselar) | Puede imitar una masa selar, especialmente si está parcialmente trombosado. Puede causar parálisis de NC (III, IV, V, VI debido a la ubicación en el seno cavernoso), dolor de cabeza. Riesgo de ruptura (HSA). | La RM muestra un vacío de flujo característico (si está permeable), señal laminada de trombo (si está presente), realce de la pared. MRA/ATC/ASD confirma el diagnóstico y delinea la anatomía vascular. |
| Cordoma / Condrosarcoma | Tumores malignos raros o localmente agresivos de la base del cráneo que surgen de restos notocordales (cordoma) o cartílago (condrosarcoma). Pueden extenderse a la región selar/supraselar desde el clivus o el ápice petroso. A menudo causan una gran destrucción ósea y parálisis de NC. | La TC muestra destrucción ósea. La RM muestra una masa expansiva localmente destructiva que se realza, a menudo hiperintensa en T2 (esp. cordoma debido a las células fisalíforas). Típicamente centrados en el clivus o unión petroclival. |
| Germinoma (Supraselar) | Raro tumor de células germinales, usualmente en niños/adultos jóvenes. Tríada clásica: DI, pérdida de visión, hipopituitarismo. Puede involucrar la región pineal de forma simultánea ("tumores dobles de la línea media"). | Masa supraselar con realce homogéneo, que a menudo afecta el quiasma óptico, tallo/hipotálamo. Los marcadores tumorales en LCR/suero (AFP, β-hCG) pueden estar elevados (aunque a menudo son normales en el germinoma puro). Altamente radiosensible. |
El diagnóstico suele involucrar Imágenes por Resonancia Magnética (RM) de la glándula pituitaria, el estándar de oro para visualizar adenomas (microadenomas, macroadenomas, incluidos los prolactinomas), evaluar su tamaño, extensión, relación con las estructuras circundantes como el quiasma óptico y los senos cavernosos, y ayudar a diferenciarlos de otras lesiones selares.
El abordaje diagnóstico combina la evaluación clínica, pruebas hormonales y estudios de imagen:
- Sospecha clínica: Basada en los síntomas de exceso hormonal (p. ej., características de acromegalia, signos cushingoides, galactorrea/amenorrea), efecto de masa (cambios visuales, dolor de cabeza, parálisis de nervios craneales), hipopituitarismo (fatiga, hipogonadismo, intolerancia al frío, etc.) o hallazgo incidental en un estudio de imagen.
- Evaluación hormonal: La medición de los niveles basales de las hormonas de la hipófisis anterior y de los órganos diana es esencial para identificar el exceso o la deficiencia de hormonas. Las pruebas clave incluyen:
- Prolactina (PRL): Los niveles elevados sugieren hiperprolactinemia. Los niveles marcadamente elevados (>200-250 ng/mL o mcg/L) son muy sugerentes de un prolactinoma (especialmente macroadenoma). Los niveles moderadamente elevados (25-150 ng/mL) requieren una evaluación de otras causas (medicamentos, hipotiroidismo, efecto de compresión del tallo, etc.). Debe verificarse el "efecto gancho" (hook effect) con una muestra diluida si hay un macroadenoma presente y los niveles de PRL son inesperadamente bajos o normales [24].
- Hormona del Crecimiento (GH) / Factor de crecimiento similar a la insulina tipo 1 (IGF-1): La IGF-1 elevada para la edad y el sexo es la mejor prueba de detección de la acromegalia. El diagnóstico se confirma mediante la falta de supresión de la GH (< 0,4 o < 1 ng/mL dependiendo del ensayo) durante una prueba de tolerancia oral a la glucosa (PTOG). Los niveles basales de GH son menos confiables debido a la pulsatilidad.
- ACTH / Cortisol: Las pruebas de detección del síndrome de Cushing incluyen pruebas como el cortisol salival nocturno, cortisol libre en orina de 24 horas (CLU) o la prueba de supresión con dosis baja (1 mg) de dexametasona durante la noche. Si es positivo, los niveles plasmáticos de ACTH ayudan a diferenciar las causas dependientes de ACTH (hipófisis/ectópico) frente a las independientes de ACTH (suprarrenales). Las pruebas de supresión con dosis altas de dexametasona y/o las pruebas de estimulación con CRH, a veces combinadas con el cateterismo de los senos petrosos inferiores (IPSS), ayudan a confirmar el origen pituitario (enfermedad de Cushing) [4].
- TSH / T4 libre/T3: La TSH elevada o inapropiadamente normal con niveles elevados de T4/T3 libre sugiere un raro TSH-oma. Las pruebas también detectan el hipotiroidismo primario (TSH alta, FT4 baja), una causa potencial de hiperprolactinemia secundaria.
- LH / FSH / Testosterona (hombres) / Estradiol (mujeres): Evalúa la función gonadal. Los niveles bajos indican hipogonadismo (secundario si la LH/FSH también son bajas/normales). La subunidad alfa elevada es común en los adenomas gonadotropos.
- Evaluación de Hipopituitarismo: Si el efecto de masa o los síntomas sugieren deficiencia, se prueba el cortisol matutino basal y ACTH, TSH y T4 libre, LH/FSH y testosterona/estradiol (según corresponda), e IGF-1 (para el eje GH). Pruebas de estimulación dinámicas (p. ej., prueba de estimulación con ACTH, prueba de tolerancia a la insulina - estándar de oro pero riesgoso, prueba GHRH-arginina) pueden ser necesarias para confirmar deficiencias sutiles. Se debe evaluar la existencia de Diabetes Insípida (DI) si hay afectación de la hipófisis posterior (poliuria, polidipsia, osmolalidad del suero/orina).
- Imágenes:
- RM de hipófisis: Con y sin contraste de gadolinio, utilizando cortes finos dedicados (≤3 mm) a través de la silla en planos coronal y sagital. Las secuencias dinámicas con contraste son particularmente útiles para identificar microadenomas, que a menudo aparecen como áreas focales de realce retardado en comparación con la adenohipófisis normal que se realza rápidamente. La RM define con precisión el tamaño del tumor, la relación con el quiasma óptico, la invasión del seno cavernoso (clasificación de Knosp) y la desviación del tallo [25].
- Tomografía Computarizada (TC): Menos sensible para los adenomas en sí, pero mejor para mostrar la erosión ósea del piso de la silla turca o los procesos clinoides, así como las calcificaciones (más comunes en los craneofaringiomas). Es útil si la RM está contraindicada.
- Evaluación Oftalmológica: Es obligatoria la realización de pruebas formales del campo visual (habitualmente campimetría automatizada, p. ej., campo visual de Humphrey) si un adenoma está cerca del quiasma óptico o lo comprime (macroadenomas), incluso si el paciente no reporta síntomas visuales. La evaluación de la agudeza visual y de la apariencia del nervio óptico (oftalmoscopia para palidez/atrofia o papiledema) también es un procedimiento estándar.
- Diagnóstico Diferencial de Hiperprolactinemia: Si se encuentra una elevación de prolactina, se deben descartar sistemáticamente otras causas antes de diagnosticar un prolactinoma, especialmente si los niveles solo son moderadamente altos (<150 ng/mL) o si las imágenes son normales o dudosas. Hay que considerar:
- Causas fisiológicas: Embarazo, lactancia, estrés, sueño (verificar niveles matutinos), ejercicio, estimulación del pezón, relaciones sexuales.
- Medicamentos: Revisar la medicación actual (antipsicóticos - esp. risperidona, típicos; antidepresivos - ISRS, ATC; antieméticos - metoclopramida, domperidona; antihipertensivos - verapamilo, metildopa; opioides; estrógenos). Suspender el medicamento sospechoso si es posible y volver a medir la PRL después de ~3 días (o según la semivida más larga).
- Condiciones sistémicas: Hipotiroidismo primario (verificar TSH/FT4), enfermedad renal crónica (verificar creatinina), cirrosis hepática grave, síndrome de ovario poliquístico (SOP), lesiones en la pared torácica (herpes zóster, cicatrices de cirugía, piercings).
- Efecto del tallo pituitario: La compresión del tallo pituitario por tumores no secretores de prolactina (NFPAs, craneofaringiomas, meningiomas) o lesiones inflamatorias interrumpe el suministro de dopamina desde el hipotálamo a las células lactotropas, lo que provoca una hiperprolactinemia leve o moderada (generalmente <150 ng/mL). Las imágenes son fundamentales.
- Macroprolactinemia: Presencia de complejos de prolactina grandes, biológicamente menos activos; requiere pruebas de laboratorio específicas (precipitación con PEG); generalmente es clínicamente insignificante.
Los niveles normales de prolactina sérica varían levemente dependiendo del laboratorio y del ensayo, pero por lo general son (rangos aproximados):
Grupo de Pacientes |
Rango Normal Aproximado de Prolactina (ng/mL o mcg/L) |
(Unidades SI Aproximadas: mIU/L) |
|---|---|---|
Niños (pre-púberes) |
Variable, generalmente < 15-20 | ~ < 300-400 |
Hombres Adultos |
< 15-20 | ~ < 300-400 |
Mujeres Adultas no embarazadas |
< 25-30 | ~ < 500-600 |
Mujeres Embarazadas |
Significativamente elevado, aumenta durante el embarazo (p. ej., hasta 200-400+ ng/mL) | ~ hasta 4000-8000+ mIU/L |
Posparto (lactancia) |
Elevado, los niveles fluctúan con la lactancia, disminuyen gradualmente si no amamantan | Variable, puede ser muy alto inicialmente |
El factor de conversión es aproximado (1 ng/mL ≈ 21 mIU/L basado en estándares de la OMS, pero varía según el ensayo).

Tratamiento del Adenoma Hipofisario y Síndrome de Hiperprolactinemia
Los objetivos del tratamiento para los adenomas hipofisarios son normalizar los niveles hormonales (si hay exceso), aliviar los síntomas por efecto de masa (pérdida visual, dolor de cabeza, parálisis de nervios craneales), preservar o restaurar la función pituitaria normal, y prevenir la recurrencia del tumor. El enfoque específico depende en gran medida del tipo de adenoma (funcionante frente a no funcionante), el tamaño (micro frente a macro), la hormona secretada, la presencia y gravedad del efecto de masa, la edad del paciente, los objetivos reproductivos y las comorbilidades [26, 2, 4, 23].
- Prolactinomas (Hiperprolactinemia):
- Terapia Médica (Primera línea): Los agonistas dopaminérgicos (DAs) son altamente efectivos y el tratamiento inicial preferido para casi todos los prolactinomas, incluidos los grandes macroadenomas que causan defectos en el campo visual (a menos que la pérdida de visión sea grave/rápidamente progresiva).
- Cabergolina: Generalmente se prefiere debido a su mayor eficacia en normalizar la prolactina y encoger los tumores, una vida media más larga que permite una dosis una o dos veces por semana, y una mejor tolerabilidad en comparación con la bromocriptina.
- Bromocriptina: Agente más antiguo, requiere dosificación diaria, se asocia más con náuseas/hipotensión postural, pero tiene un historial de seguridad más largo, especialmente durante el embarazo.
- Cirugía (Intervención neuroquirúrgica - Cirugía Transesfenoidal): Indicada principalmente para pacientes con intolerancia o resistencia a los agonistas de la dopamina, aquellos con apoplejía hipofisaria en un prolactinoma, raros casos con grandes componentes quísticos o hemorrágicos que no responden a los DAs, o a veces mujeres que desean embarazarse y no toleran o desean evitar los DAs. Las tasas de curación son más altas para los microadenomas (~70-90%) que para los macroadenomas invasivos (~30-50%).
- Radioterapia (Radiocirugía Estereotáctica o RT fraccionada): Reservada para casos raros de prolactinomas invasivos/agresivos que son resistentes o recurren tras la máxima terapia médica y quirúrgica. Se usa primordialmente para el control tumoral, dado que el efecto en los niveles de prolactina es lento y a menudo incompleto; alto riesgo de hipopituitarismo.
- Terapia Médica (Primera línea): Los agonistas dopaminérgicos (DAs) son altamente efectivos y el tratamiento inicial preferido para casi todos los prolactinomas, incluidos los grandes macroadenomas que causan defectos en el campo visual (a menos que la pérdida de visión sea grave/rápidamente progresiva).
- Acromegalia (Adenomas secretores de GH):
- Cirugía (Generalmente Primera línea): La cirugía transesfenoidal es el tratamiento primario destinado a lograr la resección completa y la cura bioquímica (normalización de IGF-1 según edad/sexo y supresión de GH durante PTOG). Las tasas de curación dependen del tamaño del tumor, invasividad y la experiencia del cirujano.
- Terapia Médica: Se utiliza para enfermedad persistente tras la cirugía, pacientes no aptos para cirugía, o a veces de forma preoperatoria para mejorar los resultados quirúrgicos. Las opciones incluyen:
- Ligandos de los Receptores de Somatostatina (SRLs): De primera generación (octreotida LAR, lanreotida Autogel) normalizan la IGF-1/GH en ~50-60% y pueden reducir los tumores. De segunda generación (pasireotida LAR) puede ser eficaz si los SRLs de primera generación fallan, pero conlleva mayor riesgo de hiperglucemia.
- Antagonista del Receptor de GH (Pegvisomant): Altamente eficaz en la normalización de la IGF-1 (~80-90%) bloqueando la acción de la GH en el receptor; no reduce el tamaño del tumor y requiere monitorización de la función hepática y el tamaño tumoral.
- Agonistas Dopaminérgicos (Cabergolina): Menos eficaces, normalizan la IGF-1 solo en un ~10-30%, se usan principalmente como terapia combinada, especialmente si el tumor cosecreta prolactina.
- Radioterapia (Estereotáctica o Fraccionada): Eficaz para controlar el crecimiento del tumor y disminuir lentamente los niveles de GH/IGF-1 en pacientes con enfermedad residual o recurrente que no responden a la terapia médica. Alto riesgo de hipopituitarismo diferido.
- Enfermedad de Cushing (Adenomas secretores de ACTH):
- Cirugía (Primera línea): La adenomectomía selectiva transesfenoidal es el tratamiento de elección, buscando la remisión bioquímica y a la vez preservando la función pituitaria normal. Las tasas de remisión son de ~70-90% para microadenomas en centros con experiencia [27].
- Terapia Médica: Se emplea de manera preoperatoria para controlar el hipercortisolismo severo, en el posoperatorio para enfermedad persistente/recurrente mientras se aguardan los efectos de la RT, o como terapia principal si la cirugía fracasa o está contraindicada. Los fármacos actúan en la esteroidogénesis suprarrenal (ketoconazol, metirapona, etomidato, osilodrostat), la secreción pituitaria de ACTH (pasireotida, cabergolina), o bloquean el receptor de glucocorticoides (mifepristona).
- Radioterapia (Estereotáctica o Fraccionada): Efectiva para la enfermedad persistente o recurrente tras la cirugía, pero la remisión bioquímica tarda (meses o años).
- Adrenalectomía Bilateral: Proporciona una curación definitiva del hipercortisolismo si las terapias dirigidas a la hipófisis fallan, pero da como resultado una insuficiencia suprarrenal permanente que requiere tratamiento sustitutivo vitalicio con glucocorticoides y mineralocorticoides, y conlleva el riesgo del síndrome de Nelson (crecimiento progresivo/pigmentación por el tumor pituitario secretor de ACTH).
- TSH-omas (raro):
- Cirugía (Primera línea): La resección transesfenoidal es el tratamiento preferido.
- Terapia Médica: Los ligandos de los receptores de somatostatina (octreotida, lanreotida) son a menudo efectivos para suprimir la secreción de TSH y encoger los tumores de forma preoperatoria o para la enfermedad residual. Los antitiroideos (metimazol, PTU) manejan los síntomas de hipertiroidismo pero no tratan el tumor.
- Radioterapia: Para tumores residuales/recurrentes que no responden a otras terapias.
- Adenomas Hipofisarios No Funcionantes (NFPAs): Consulte la sección a continuación.
Las decisiones de manejo deben ser individualizadas y a menudo requieren un enfoque de equipo multidisciplinario que incluye endocrinólogos, neurocirujanos, neurorradiólogos, oftalmólogos y en ocasiones radiooncólogos.
Tratamiento de los Adenomas Hipofisarios No Funcionantes (NFPAs)
Dado que los NFPAs normalmente no causan síndromes por exceso de hormonas, el tratamiento está indicado principalmente cuando causan efecto de masa (deterioro visual, parálisis de los nervios craneales, dolores de cabeza clínicamente significativos atribuibles al tumor) o hipopituitarismo, o si demuestran un crecimiento significativo en las imágenes en serie [22, 23].
- NFPAs incidentales asintomáticos:
- Microadenomas (<10mm): Generalmente se manejan de manera conservadora con vigilancia periódica por RM (p. ej., RM a 1 año, luego cada 1-2 años, luego con menos frecuencia si está estable) y evaluación endocrina básica (detección de hipopituitarismo e hiperprolactinemia debida al efecto sobre el tallo). La mayoría (~90-95%) no crece de manera significativa [8].
- Macroadenomas (≥10mm) sin efecto de masa (sin déficits visuales, función pituitaria normal, sin apoyarse sobre el quiasma): El manejo se individualiza. Las opciones incluyen un seguimiento cercano con RM (p. ej., a los 6 meses, luego de manera anual si está estable) y evaluaciones endocrinas/visuales regulares, o la consideración de una cirugía, especialmente si el tumor es grande o se encuentra muy próximo al quiasma óptico, dada la posibilidad de futuro crecimiento o complicaciones. La intervención está indicada si hay un crecimiento tumoral documentado, desarrollo de síntomas (pérdida visual, hipopituitarismo), o si el tumor llega a contactar/comprimir el quiasma óptico [23].
- NFPAs sintomáticos (que causan efecto de masa o hipopituitarismo):
- Intervención Quirúrgica (Primera línea): La cirugía transesfenoidal es el tratamiento principal. Los objetivos fundamentales son la descompresión del aparato óptico para mejorar o preservar la visión y la máxima extirpación segura del tumor para aliviar el efecto de masa y permitir potencialmente la recuperación de la función hipofisaria (menos probable si los déficits son de larga data). En la actualidad se emplean comúnmente abordajes endoscópicos endonasales. La resección completa es frecuentemente difícil para los tumores grandes o invasivos (p. ej., hacia el seno cavernoso).
- Radioterapia (Radiocirugía Estereotáctica - SRS o RT Fraccionada): Indicada principalmente como tratamiento coadyuvante para el tumor residual significativo tras la cirugía (en particular si es un volumen residual que está creciendo o es voluminoso) o para tumores recurrentes. Rara vez se emplea la RT primaria a no ser que la cirugía esté contraindicada. La RT ofrece excelentes tasas de control tumoral a largo plazo (>90%), pero no reduce inmediatamente el efecto de masa y conlleva un importante riesgo retardado (~30-60% en 10 años) de hipopituitarismo inducido por la radiación [28].
- Terapia Médica: No existe actualmente una terapia médica eficaz demostrada para reducir el tamaño de los NFPAs o evitar su crecimiento. Se han investigado los agonistas de la dopamina o los análogos de la somatostatina, pero resultan ser por lo general ineficaces para los NFPAs gonadotropos típicos.
- Manejo del Hipopituitarismo: Las deficiencias hormonales identificadas antes o después del tratamiento precisan un tratamiento sustitutivo fisiológico adecuado de por vida (p. ej., hidrocortisona para la insuficiencia suprarrenal con necesidad de cobertura de dosis de estrés; levotiroxina para el hipotiroidismo secundario; testosterona/estrógeno +/- progesterona para el hipogonadismo; de manera potencial, sustitución de GH en adultos con una deficiencia confirmada e importante impacto sobre la calidad de vida o los parámetros metabólicos; DDAVP para la diabetes insípida si existe).
- Seguimiento a Largo Plazo: Es fundamental luego del tratamiento (cirugía +/- radiación) para la totalidad de los macroadenomas. Incluye la monitorización por posible recurrencia o recrecimiento del tumor (RM seriada, la frecuencia dependerá del tumor residual o tipo de tratamiento), la evaluación habitual de la función endocrina (pruebas anuales por hipopituitarismo, también luego de la RT), y vigilar la situación visual (campos visuales de haber antecedentes de compresión). Las tasas de recurrencia tras realizar sólo la intervención quirúrgica pueden ser considerables con el tiempo (10-50% según la duración de la observación y el tumor residual), y exigen plantearse la necesidad de someter a radioterapia coadyuvante cuando hay un tumor residual preocupante.
![]() ¡Atención! Los adenomas hipofisarios requieren diagnóstico y tratamiento especializados por un equipo multidisciplinario. Síntomas como cambios en la visión, dolores de cabeza persistentes, desequilibrios hormonales (alteraciones menstruales, lactancia, cambios físicos no explicados, disfunción sexual), o fatiga excesiva justifican una evaluación por un médico, pudiendo derivar a un endocrinólogo y un neurocirujano.
¡Atención! Los adenomas hipofisarios requieren diagnóstico y tratamiento especializados por un equipo multidisciplinario. Síntomas como cambios en la visión, dolores de cabeza persistentes, desequilibrios hormonales (alteraciones menstruales, lactancia, cambios físicos no explicados, disfunción sexual), o fatiga excesiva justifican una evaluación por un médico, pudiendo derivar a un endocrinólogo y un neurocirujano.
Referencias
- Melmed S. Pituitary-Tumor Endocrinopathies. N Engl J Med. 2020 Mar 5;382(10):937-950. doi: 10.1056/NEJMra1810772
- Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011 Feb;96(2):273-88. doi: 10.1210/jc.2010-1692
- Katznelson L, Laws ER Jr, Melmed S, et al; Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Nov;99(11):3933-51. doi: 10.1210/jc.2014-2700
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008 May;93(5):1526-40. doi: 10.1210/jc.2008-0125
- Beck-Peccoz P, Persani L. Thyrotropin-secreting pituitary adenomas. N Engl J Med. 1996;334(11):712-713. doi: 10.1056/NEJM199603143341107
- Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2011 Jun;14(2):111-8. doi: 10.1007/s11102-011-0300-1
- Molitch ME. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 1997 Mar;26(1):1-17. doi: 10.1016/s0889-8529(05)70239-7
- Fernández-Balsells MM, Murad MH, Barwise A, et al. Clinical management of nonfunctioning pituitary adenomas (NFPAs): a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011 Apr;96(4):905-15. doi: 10.1210/jc.2010-1921
- Chapter on Pituitary Imaging. In: Osborn AG, Salzman KL, Jhaveri MD, et al. Osborn's Brain. 2nd ed. Elsevier; 2018.
- Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. 2002 Feb;2(2):83-94. doi: 10.1038/nrc760
- Vierimaa O, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006 May 26;312(5777):1228-30. doi: 10.1126/science.1126100
- Reincke M, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. 2015 Jan;47(1):31-8. doi: 10.1038/ng.3166
- Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936 Mar;12(2):205-16.1.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004 Aug 1;101(3):613-9. doi: 10.1002/cncr.20412
- Hall WA, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994 May 15;120(10):817-20. doi: 10.7326/0003-4819-120-10-199405150-00001
- Ciccarelli A, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Varese, Italy. J Clin Endocrinol Metab. 2001 Feb;86(2):720-5. doi: 10.1210/jcem.86.2.7204
- Fernandez A, et al. Prevalence of pituitary adenomas: a community-based study in Olmsted County, Minnesota, 1993-1997. Arch Intern Med. 2000 Jan 10;160(1):78-81. doi: 10.1001/archinte.160.1.78
- Daly AF, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006 Dec;91(12):4756-69. doi: 10.1210/jc.2006-1668
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009 Mar;119(3):429-38. doi: 10.1172/JCI38090
- Nieman LK. Cushing's syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015 Oct;173(4):M33-8. doi: 10.1530/EJE-15-0464
- Elster AD, et al. Pituitary gland: MR imaging of physiologic hypertrophy in adolescence. Radiology. 1990;174(3 Pt 1):681-5. doi: 10.1148/radiology.174.3.2305044
- Molitch ME. Nonfunctioning pituitary adenomas. Pituitary. 2005;8(3-4):213-20. doi: 10.1007/s11102-006-8512-6
- Freda PU, Beckers AM, Katznelson L, et al; Endocrine Society. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011 Apr;96(4):894-904. doi: 10.1210/jc.2010-1048
- Frieze TW, et al. Hook effect in prolactinomas: case report and review of literature. Endocr Pract. 2006;12(3):296-303. doi: 10.4158/EP.12.3.296
- Bonneville F, et al. Radiology of pituitary adenomas: an update. Radiol Clin North Am. 2012 Nov;50(6):995-1013. doi: 10.1016/j.rcl.2012.08.005
- Melmed S, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509-17. doi: 10.1210/jc.2008-2421
- Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818
- Sheehan JP, et al. Radiosurgery for nonfunctioning pituitary adenomas: a systematic review. J Neurosurg. 2013 Aug;119(2):446-56. doi: 10.3171/2013.4.JNS121833
Ver también
- Anatomía del sistema nervioso
- Infección del sistema nervioso central:
- Absceso cerebral (lobar, cerebeloso)
- Granuloma eosinofílico, histiocitosis de células de Langerhans (HCL), síntoma de Hennebert
- Absceso cerebral epidural
- Complicaciones intracraneales asociadas a sinusitis
- Complicaciones intracraneales otógenas
- Complicaciones oftálmicas asociadas a sinusitis
- Meningitis bacteriana otógena
- Absceso cerebral subdural
- Tromboflebitis supurativa del seno sigmoideo
- Quiste coloide del tercer ventrículo cerebral
- Aracnoiditis adhesiva cerebral y espinal
- Degeneración corticobasal (atrofia cerebral focalizada)
- Encefalopatía
- Dolor de cabeza, migraña
- Lesión cerebral traumática (conmoción, contusión, hemorragia cerebral, daño axonal difuso)
- Hipertensión intracraneal e hidrocefalia
- Enfermedad de Parkinson
- Microadenoma hipofisario, macroadenoma y adenomas no funcionantes (NFPAs), síndrome de hiperprolactinemia
- Fuga craneal espontánea de líquido cefalorraquídeo (LCR)