Microadénome hypophysaire, macroadénome et adénomes non fonctionnels (NFPAs), syndrome d'hyperprolactinémie
- Aperçu de l'adénome hypophysaire (microadénome, macroadénome) et du syndrome d'hyperprolactinémie
- Microadénomes hypophysaires :
- Macroadénomes hypophysaires :
- Adénomes hypophysaires non fonctionnels (NFPAs)
- Diagnostic de l'adénome hypophysaire et du syndrome d'hyperprolactinémie
- Traitement de l'adénome hypophysaire et du syndrome d'hyperprolactinémie
- Traitement des adénomes hypophysaires non fonctionnels (NFPAs)
- Références
Aperçu de l'adénome hypophysaire (microadénome, macroadénome) et du syndrome d'hyperprolactinémie
Les adénomes hypophysaires sont typiquement des tumeurs bénignes (non cancéreuses) provenant des cellules productrices d'hormones du lobe antérieur de l'hypophyse (adénohypophyse), située à la base du cerveau dans une structure osseuse appelée selle turcique [1]. Ces adénomes sont classés en fonction de leur taille (microadénome par rapport à macroadénome) et de leur activité hormonale (fonctionnels par rapport à non fonctionnels).
Le syndrome d'hyperprolactinémie fait référence aux conséquences cliniques de niveaux de prolactine, une hormone normalement impliquée dans la lactation, élevés de façon persistante. Les prolactinomes (adénomes hypophysaires sécrétant de la prolactine) sont le type le plus courant d'adénome hypophysaire fonctionnel et une cause fréquente d'hyperprolactinémie [2]. Chez les femmes, les symptômes incluent généralement une galactorrhée (production de lait inappropriée), des irrégularités menstruelles (oligoménorrhée ou aménorrhée) et une infertilité. Chez les hommes, l'hyperprolactinémie entraîne souvent une baisse de la libido, une dysfonction érectile, une infertilité due à l'oligospermie, une gynécomastie (développement des seins) et, moins fréquemment, une galactorrhée. Une prolactine élevée inhibe la sécrétion de la gonadolibérine (GnRH) par l'hypothalamus, entraînant un hypogonadisme secondaire.

Les symptômes liés aux adénomes hypophysaires proviennent soit d'un excès de sécrétion hormonale (dans les adénomes fonctionnels), soit de la taille physique de la tumeur et de la pression sur les structures environnantes (effet de masse), particulièrement avec les macroadénomes. Les syndromes d'excès hormonal incluent :
Hormone en excès |
Condition résultante & Principaux problèmes de santé |
|---|---|
| Prolactine (PRL) |
Hyperprolactinémie : - Femmes : Dysfonctionnement menstruel (oligoménorrhée, aménorrhée), galactorrhée, infertilité, diminution de la densité osseuse. - Hommes : Baisse de la libido, dysfonction érectile, infertilité, gynécomastie (moins courant), diminution de la densité osseuse. [2] |
| Hormone de croissance (GH) |
Acromégalie (adultes) / Gigantisme (enfants) : - Élargissement des mains, des pieds, des traits du visage (mâchoire, front). - Gonflement des tissus mous, transpiration excessive. - Arthrose, syndrome du canal carpien. - Apnée du sommeil. - Hypertension, cardiomyopathie. - Diabète sucré ou intolérance au glucose. - Risque accru de polypes/cancer colorectal. [3] |
| Hormone adrénocorticotrope (ACTH) |
Maladie de Cushing (Syndrome de Cushing dépendant de l'hypophyse) : - Obésité centrale (graisse tronculaire, faciès lunaire, bosse de bison), peau fine, ecchymoses faciles, vergetures pourpres. - Faiblesse musculaire (myopathie proximale), ostéoporose. - Changements d'humeur (anxiété, dépression, psychose). - Hypertension, diabète sucré. - Sensibilité accrue aux infections. - Irrégularités menstruelles (femmes). [4] |
| Hormone thyréostimulante (TSH) |
Adénome thyréotrope (TSH-ome, rare) : - Hyperthyroïdie secondaire : Les symptômes incluent intolérance à la chaleur, transpiration, tachycardie/palpitations (arythmies comme la fibrillation auriculaire), tremblements, anxiété/irritabilité, perte de poids involontaire malgré un appétit accru, goitre. Caractérisé par une TSH anormalement normale ou élevée avec des niveaux élevés de T4/T3 libres. [5] |
| Gonadotrophines (LH, FSH) |
Adénome gonadotrope (souvent cliniquement non fonctionnel) : - Sécrètent généralement des fragments hormonaux inactifs ou intacts (par ex., sous-unité alpha, bêta-FSH, moins fréquemment FSH/LH intactes). - Peuvent rarement causer une hyperstimulation ovarienne (femmes) ou un élargissement testiculaire/puberté précoce (hommes). - Se présentent le plus souvent en raison de l'effet de masse ou sont découverts de manière fortuite. [6] |
Les adénomes hypophysaires non fonctionnels (NFPAs) ne sécrètent pas de quantités cliniquement significatives d'hormones actives, bien que beaucoup produisent des sous-unités hormonales détectables par des tests spéciaux. Ils provoquent généralement des symptômes dus à l'effet de masse lorsqu'ils deviennent des macroadénomes, comprimant les structures adjacentes comme le chiasma optique (causant des déficits du champ visuel, classiquement une hémianopsie bitemporale), les nerfs crâniens dans le sinus caverneux (causant une vision double) ou le tissu hypophysaire normal (causant un hypopituitarisme - déficit en une ou plusieurs hormones hypophysaires). Les maux de tête sont également fréquents en raison de l'étirement de la dure-mère ou de l'augmentation de la pression sellaire.
Microadénomes hypophysaires
Description des microadénomes hypophysaires
Un microadénome hypophysaire est défini comme un adénome hypophysaire ayant un diamètre maximal de moins de 10 millimètres (moins de 1 cm) [7]. Ces tumeurs sont confinées à l'intérieur de la selle turcique et ne causent généralement pas de symptômes d'effet de masse comme la perte de vision. Les microadénomes peuvent être soit fonctionnels (sécrétant des hormones), soit non fonctionnels. Les microadénomes fonctionnels provoquent des symptômes liés à l'hormone spécifique qu'ils surproduisent (par ex., prolactinome causant une hyperprolactinémie, sécrétant de l'ACTH causant la maladie de Cushing, sécrétant de la GH causant l'acromégalie).
Cependant, de nombreux microadénomes sont cliniquement non fonctionnels et sont découverts de manière fortuite lors d'une imagerie cérébrale (Imagerie par résonance magnétique - IRM ou TDM) réalisée pour d'autres raisons (par ex., maux de tête, vertiges, traumatisme). On les appelle souvent des « incidentalomes » hypophysaires. Bien que la grande majorité soit des découvertes bénignes ne nécessitant qu'une surveillance, même les petits microadénomes fortuits peuvent parfois causer de subtils déséquilibres hormonaux (Cushing subclinique, hyperprolactinémie légère) ou, rarement (< 10 % sur plusieurs années), croître avec le temps [8].
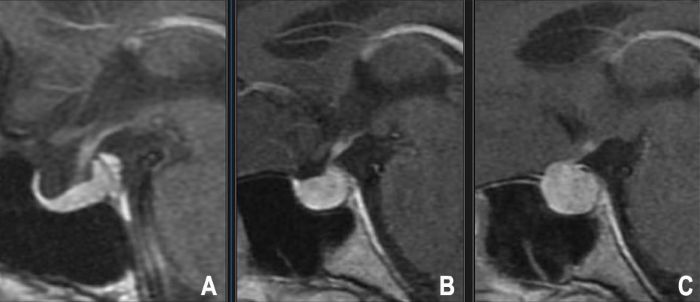
L'IRM avec contraste au gadolinium, utilisant particulièrement des séquences dynamiques avec contraste et des coupes fines à travers la fosse hypophysaire, est la modalité d'imagerie de choix pour détecter les microadénomes. Ils apparaissent souvent comme de petites lésions focales qui se rehaussent moins ou plus tardivement que le tissu de la glande hypophysaire normale environnante (typiquement hypointenses sur les premières images T1 post-contraste) [9].
Les microadénomes cliniquement significatifs sont le plus souvent des prolactinomes, suivis par les adénomes sécrétant de l'ACTH causant la maladie de Cushing, et les adénomes sécrétant de la GH causant l'acromégalie. Les microadénomes non fonctionnels sont souvent des adénomes gonadotropes par immunohistochimie mais manquent d'excès hormonal clinique. Le diagnostic différentiel en imagerie inclut les kystes de la poche de Rathke, l'hyperplasie hypophysaire, les métastases ou les processus inflammatoires (hypophysite), bien que ceux-ci présentent souvent des caractéristiques ou un contexte clinique distincts.
Physiopathologie des microadénomes hypophysaires
Les adénomes hypophysaires, y compris les microadénomes, sont généralement considérés comme des néoplasmes monoclonaux, ce qui signifie qu'ils proviennent de l'expansion clonale d'une seule cellule mutée de l'adénohypophyse [10]. Les mutations initiales spécifiques et les événements génétiques/épigénétiques ultérieurs conduisant à la formation de l'adénome sont complexes et variés, mais plusieurs facteurs et voies sont impliqués.
Des syndromes génétiques germinaux spécifiques prédisposent les individus aux adénomes hypophysaires : la Néoplasie endocrinienne multiple de type 1 (NEM1), causée par des mutations dans le gène suppresseur de tumeur MEN1, implique fréquemment des prolactinomes ou des adénomes sécrétant de la GH. Le syndrome de McCune-Albright, dû à des mutations activatrices en mosaïque dans le gène GNAS codant pour la sous-unité alpha de la protéine Gs, entraîne souvent des adénomes sécrétant de la GH (somatotropinomes). Le complexe de Carney (mutations PRKAR1A) peut inclure des adénomes hypophysaires (typiquement sécrétant de la GH). Les mutations dans le gène AIP (protéine interagissant avec le récepteur des hydrocarbures aryliques) prédisposent aux adénomes hypophysaires isolés familiaux (FIPA), en particulier les tumeurs sécrétant de la GH ou de la prolactine, se présentant souvent à un jeune âge et étant plus grandes/plus agressives [11].
Les adénomes sporadiques (non familiaux) impliquent également des altérations génétiques et épigénétiques somatiques. Des mutations somatiques activatrices dans GNAS se trouvent dans environ 40 pour cent des adénomes sporadiques sécrétant de la GH. Les mutations de USP8 sont courantes dans les adénomes sécrétant de l'ACTH [12]. D'autres mutations somatiques et modifications épigénétiques affectant les régulateurs du cycle cellulaire (par ex., perte de fonction de CDKN1B/p27, altérations des voies p53, RB), les voies des facteurs de croissance (par ex., surexpression de FGFR4), et les facteurs de transcription spécifiques à l'hypophyse contribuent à la tumorigenèse et à la production hormonale. La perte d'hétérozygotie à certains emplacements chromosomiques (par ex., 11q13 où réside MEN1) est également observée.
Les prolactinomes, le type le plus courant, proviennent des cellules lactotropes. Les corticotropinomes causant la maladie de Cushing proviennent des cellules corticotropes. Les somatotropinomes causant l'acromégalie proviennent des cellules somatotropes. Les gonadotropinomes proviennent généralement des cellules gonadotropes et sécrètent souvent des sous-unités hormonales inactives (sous-unité alpha, bêta-FSH, bêta-LH). Les thyréotropinomes causant une hyperthyroïdie proviennent des cellules thyréotropes et sont rares.
La plupart des adénomes cliniquement non fonctionnels sont immunopositifs pour les sous-unités des gonadotrophines (FSH, LH, sous-unité alpha) mais ne provoquent pas de syndromes cliniques d'excès hormonal. Certains peuvent être des adénomes à "cellules nulles" sans coloration hormonale spécifique, ou des adénomes corticotropes/somatotropes silencieux.
Épidémiologie des microadénomes hypophysaires
Fréquence des microadénomes hypophysaires
Les microadénomes hypophysaires sont des découvertes fortuites étonnamment courantes. Les études d'autopsie ont rapporté des adénomes microscopiques ou de petite taille (souvent < 3 mm) chez jusqu'à 27 pour cent des individus sans maladie hypophysaire connue [13, 14]. Les études d'imagerie haute résolution (IRM) détectent des incidentalomes hypophysaires (dont la plupart sont des microadénomes < 10 mm) chez environ 10 à 20 pour cent de la population générale subissant des scanners cérébraux pour des raisons non liées [15].
La grande majorité de ces microadénomes découverts fortuitement sont non fonctionnels et restent stables ou croissent très lentement, sans jamais causer de symptômes cliniques. Les microadénomes fonctionnels sont beaucoup moins fréquents mais sont la cause habituelle des syndromes d'excès d'hormones hypophysaires comme l'hyperprolactinémie (le prolactinome étant le type clinique diagnostiqué le plus fréquent, prévalence estimée de ~10 à 50 pour 100 000 habitants [16]), la maladie de Cushing et l'acromegalie.
Les microadénomes hypophysaires peuvent survenir à tout âge mais sont le plus souvent diagnostiqués chez les jeunes adultes à ceux d'âge moyen (de 20 à 50 ans). Il existe une prédominance féminine pour les prolactinomes (ratio F:H ~10:1) et la maladie de Cushing (~3-5:1), tandis que les adénomes sécrétant de la GH ont une répartition par sexe à peu près égale.
Avant l'utilisation généralisée de l'IRM, l'hyperprolactinémie détectée via des tests sanguins était un indice majeur de microadénomes sous-jacents sécrétant de la prolactine, souvent invisibles. L'IRM permet désormais une visualisation directe.
La prévalence élevée des microadénomes découverts de manière fortuite par rapport à la prévalence plus faible de la maladie hypophysaire cliniquement significative ou des macroadénomes suggère que la plupart des microadénomes ne progressent pas pour devenir des tumeurs de grande taille ou symptomatiques.
Macroadénomes hypophysaires
Description des macroadénomes hypophysaires
Un macroadénome hypophysaire est défini comme un adénome hypophysaire mesurant 10 millimètres (1 cm) ou plus dans son diamètre maximum [3]. Contrairement aux microadénomes, les macroadénomes s'étendent fréquemment au-delà des limites de la selle turcique, pouvant potentiellement comprimer les structures neurales et vasculaires environnantes.
Ils peuvent être fonctionnels (sécrétant des hormones) ou non fonctionnels. Les adénomes hypophysaires non fonctionnels (NFPAs) sont le type de macroadénome le plus couramment diagnostiqué sur le plan clinique [6]. Les macroadénomes fonctionnels incluent de grands prolactinomes, des adénomes sécrétant de la GH causant l'acromégalie, et moins fréquemment, de grands adénomes sécrétant de l'ACTH (souvent associés au syndrome de Nelson après une surrénalectomie) ou des adénomes sécrétant de la TSH.
Les macroadénomes se présentent souvent avec des symptômes liés à l'effet de masse en raison de leur taille :
- Troubles visuels : La compression du chiasma optique situé au-dessus de l'hypophyse est courante lors d'une extension suprasellaire, provoquant classiquement une hémianopsie bitemporale (perte de la vision périphérique dans les deux champs temporaux externes). La compression des nerfs optiques ou des voies optiques peut causer d'autres types de perte de vision ou une diminution de l'acuité visuelle.
- Paralysies des nerfs crâniens : L'extension latérale dans le sinus caverneux peut comprimer les nerfs crâniens III (oculomoteur), IV (trochléaire), V (trijumeau - branches ophtalmique V1 et maxillaire V2) et VI (abducens), entraînant une vision double (diplopie), un affaissement des paupières (ptosis), des modifications pupillaires ou des engourdissements/douleurs au visage. Les nerfs crâniens III et VI sont le plus souvent affectés cliniquement.
- Maux de tête : Souvent non spécifiques, potentiellement dus à l'étirement de la dure-mère couvrant la selle (diaphragme sellaire) ou à l'augmentation de la pression intrasellaire.
- Hypopituitarisme : La compression et la destruction du tissu de la glande hypophysaire normale peuvent entraîner une carence en une ou plusieurs hormones de l'hypophyse antérieure (la carence en GH est souvent la plus précoce, suivie par les gonadotrophines LH/FSH causant un hypogonadisme, puis la TSH causant une hypothyroïdie secondaire, et enfin l'ACTH causant une insuffisance surrénalienne secondaire) ou, moins couramment, un dysfonctionnement de l'hypophyse postérieure (carence en hormone antidiurétique - ADH, causant un diabète insipide central, plus souvent vu lors de la compression de la tige ou de lésions d'origine postérieure).
- Apoplexie hypophysaire : Une urgence médicale aiguë causée par une hémorragie soudaine ou un infarctus au sein d'un adénome hypophysaire (beaucoup plus fréquente dans les macroadénomes que dans les microadénomes). Se présente avec un mal de tête sévère et soudain, une perte de vision rapide, des paralysies des nerfs crâniens, une altération de la conscience et souvent un hypopituitarisme aigu (nécessitant un remplacement urgent par stéroïdes).
Les macroadénomes fonctionnels causent également des symptômes liés à la surproduction d'hormones, souvent plus graves que pour les microadénomes en raison d'un fardeau tumoral plus important, comme décrit précédemment.
Physiopathologie des macroadénomes hypophysaires
La physiopathologie sous-jacente implique l'expansion clonale de cellules adénohypophysaires mutées, de manière similaire aux microadénomes. Les facteurs entraînant la progression du microadénome vers le macroadénome ou le développement de novo de macroadénomes ne sont pas entièrement élucidés, mais impliquent probablement l'accumulation d'altérations génétiques supplémentaires (par ex., mutations des gènes suppresseurs de tumeurs comme p53, instabilité chromosomique) ou épigénétiques favorisant une prolifération accrue, une diminution de l'apoptose, l'angiogenèse et parfois l'invasivité [10].
Les macroadénomes démontrent souvent une perte des mécanismes normaux de rétroaction contrôlant la sécrétion hormonale (particulièrement ceux qui sont fonctionnels). Leur croissance entraîne la compression et la destruction des cellules hypophysaires normales adjacentes, conduisant à un hypopituitarisme. L'extension vers le haut comprime le chiasma optique et potentiellement l'hypothalamus (entraînant un DI ou une dérégulation de l'appétit/température). L'extension latérale envahit le sinus caverneux, pouvant potentiellement encercler l'artère carotide interne et les nerfs crâniens III, IV, V1, V2 et VI. L'extension vers le bas peut éroder le plancher de la selle turcique dans le sinus sphénoïdal, provoquant parfois une rhinorrhée de LCR.
La base génétique implique des facteurs mentionnés pour les microadénomes, incluant les syndromes germinaux (NEM1, GNAS, AIP, complexe de Carney) et des mutations somatiques (GNAS, USP8). Des altérations additionnelles associées à des tumeurs plus grandes ou plus agressives pourraient inclure des mutations dans les régulateurs du cycle cellulaire (p53, RB), des récepteurs des facteurs de croissance (FGFR4), ou des molécules d'adhésion. Bien que bénins d'un point de vue histologique (faible taux mitotique, pas d'atypie cytologique en général), certains macroadénomes présentent des schémas de croissance invasifs vers les structures environnantes (dure-mère, os, sinus caverneux), ce qui rend une résection chirurgicale complète difficile. Les véritables carcinomes hypophysaires primaires, définis par la présence de métastases craniospinales ou systémiques, sont extrêmement rares.
Des syndromes cliniques associés comme la NEM1, le syndrome de McCune-Albright, et le complexe de Carney peuvent impliquer des macroadénomes, souvent de type sécrétant de la GH ou de la prolactine.
Épidémiologie des macroadénomes hypophysaires
Fréquence des macroadénomes hypophysaires
Bien que les microadénomes fortuits soient très fréquents, les tumeurs hypophysaires cliniquement significatives nécessitant une évaluation et un traitement potentiel le sont moins. La prévalence globale des adénomes hypophysaires cliniquement apparents est estimée à environ 78 à 94 pour 100 000 individus, soit grosso modo 1 sur 1000 [17, 18]. Les macroadénomes constituent une proportion importante de ces tumeurs cliniquement pertinentes, représentant probablement 30 à 50 % des adénomes diagnostiqués, particulièrement parmi les adénomes non fonctionnels qui se présentent souvent en raison de leur taille.
Les études en population suggèrent une incidence annuelle globale de nouveaux adénomes hypophysaires diagnostiqués d'environ 4 pour 100 000 habitants [18], avec des taux de prévalence en augmentation en raison de l'amélioration de la détection grâce à l'imagerie. L'incidence nécessitant une intervention neurochirurgicale varie d'environ 1 à 7 pour 100 000 par an, les macroadénomes causant un effet de masse ou un excès hormonal réfractaire au traitement médical constituant les principales indications de la chirurgie.
Morbidité et mortalité dans les macroadénomes hypophysaires
La morbidité associée aux macroadénomes hypophysaires provient principalement de l'effet de masse (perte de vision, paralysies des nerfs crâniens, maux de tête invalidants), du dysfonctionnement hormonal (hypopituitarisme nécessitant un remplacement à vie, ou des conséquences systémiques des syndromes d'excès hormonal comme l'acromégalie ou la maladie de Cushing), et des complications potentielles du traitement (par ex., risques chirurgicaux tels que la fuite de LCR, la méningite, la lésion vasculaire, l'aggravation de l'hypopituitarisme ; effets à long terme de la radiothérapie comme l'hypopituitarisme, les tumeurs secondaires, les changements cognitifs). La déficience visuelle, particulièrement les déficits permanents dus à un traitement différé de la compression chiasmatique, impacte significativement la qualité de vie. L'hypopituitarisme nécessite une prise en charge minutieuse et comporte ses propres risques s'il est traité de façon inadéquate (par ex., crise surrénalienne). Les complications liées à un excès hormonal non contrôlé (par ex., maladie cardiovasculaire, diabète, ostéoporose, risque accru de cancer dans l'acromégalie et la maladie de Cushing) contribuent significativement à la morbidité et à la mortalité à long terme si elles ne sont pas efficacement gérées [19, 20].
La mortalité directement liée à la nature bénigne des macroadénomes hypophysaires eux-mêmes est généralement faible. Cependant, un risque de mortalité accru est bien documenté chez les patients atteints d'acromégalie et de maladie de Cushing non contrôlées, principalement en raison des complications cardiovasculaires (hypertension, insuffisance cardiaque, accident vasculaire cérébral, arythmies), des problèmes métaboliques (diabète) et des infections (Cushing). L'apoplexie hypophysaire, bien que rare, peut être fatale si elle n'est pas reconnue et traitée promptement. Les complications liées à la chirurgie (bien que la mortalité soit faible, <1%, dans les centres expérimentés) ou les effets à long terme de la radiothérapie peuvent également y contribuer. La survie globale pour les patients avec des macroadénomes est généralement bonne grâce à une prise en charge multidisciplinaire moderne qui contrôle les niveaux hormonaux et l'effet de masse, bien que la qualité de vie puisse être affectée par des symptômes résiduels ou des effets secondaires du traitement.
Les macroadénomes hypophysaires ne montrent aucune prédilection raciale significative.
Bien que les études d'autopsie montrent une prévalence égale d'adénomes fortuits entre les sexes, les prolactinomes cliniquement diagnostiqués et les adénomes sécrétant de l'ACTH (maladie de Cushing) sont significativement plus fréquents chez les femmes en âge de procréer, possiblement en raison d'une reconnaissance plus précoce des symptômes (irrégularités menstruelles, galactorrhée). Les adénomes sécrétant de la GH (acromégalie) et les NFPAs se présentant cliniquement ont une répartition plus égale entre les sexes. Les macroadénomes hypophysaires peuvent survenir à tout âge mais sont le plus fréquemment diagnostiqués chez les adultes, l'incidence atteignant un pic entre la troisième et la sixième décennie de la vie.
Adénomes hypophysaires non fonctionnels (NFPAs)
Les adénomes hypophysaires non fonctionnels (NFPAs) sont des tumeurs hypophysaires qui ne causent pas de syndrome clinique de sursécrétion hormonale. Ils représentent environ 15 à 30 pour cent de l'ensemble des adénomes hypophysaires cliniquement diagnostiqués et sont le type le plus courant se présentant comme des macroadénomes [22]. Bien qu'ils soient qualifiés de « non fonctionnels », la coloration immunohistochimique révèle que la plupart des NFPAs proviennent des cellules gonadotropes et produisent souvent des hormones gonadotrophines (hormone folliculostimulante - FSH, hormone lutéinisante - LH) ou leurs sous-unités (particulièrement la sous-unité alpha ou la sous-unité bêta de la FSH), mais ces hormones sont typiquement inactives biologiquement, sécrétées de manière inefficace ou dans des ratios anormaux, ne causant donc pas de symptômes cliniques d'excès hormonal [6]. Certains peuvent être de vrais adénomes à « cellules nulles » ou des variantes silencieuses d'autres cellules productrices d'hormones.
Parce qu'ils ne présentent pas de syndromes d'hypersécrétion hormonale, les NFPAs se présentent généralement plus tard que les adénomes fonctionnels, souvent lorsqu'ils sont devenus suffisamment gros (macroadénomes) pour causer des symptômes dus à l'effet de masse :
- Déficits du champ visuel (typiquement une hémianopsie bitemporale due à la compression du chiasma optique).
- Maux de tête (non spécifiques, dus à l'étirement de la dure-mère ou à la pression sellaire).
- Hypopituitarisme (dû à la compression du tissu hypophysaire normal, affectant le plus souvent en premier lieu la sécrétion de GH et des gonadotrophines, suivie par la TSH et l'ACTH).
- Paralysies des nerfs crâniens (moins courantes, provenant d'une invasion du sinus caverneux, causant une diplopie ou un engourdissement facial).
- Apoplexie hypophysaire (hémorragie/infarctus aigu au sein de la tumeur).
De nombreux petits NFPAs sont découverts fortuitement (IRM ou TDM) en tant qu'incidentalomes hypophysaires. L'autopsie et les études d'imagerie suggèrent une forte prévalence de microadénomes silencieux (jusqu'à 1 personne sur 6 pouvant potentiellement en abriter un), mais la plupart demeurent petits et asymptomatiques tout au long de la vie.
Le traitement principal pour les NFPAs symptomatiques causant un effet de masse ou un hypopituitarisme progressif est la résection chirurgicale, généralement par la voie transsphénoïdale. Les objectifs principaux sont la décompression des structures environnantes (en particulier de l'appareil optique pour améliorer/préserver la vision) et le retrait de la tumeur de la manière la plus sûre et maximale possible pour réduire l'effet de masse et potentiellement permettre la récupération de la fonction hypophysaire. La prise en charge des NFPAs fortuits asymptomatiques implique une surveillance avec une imagerie régulière et des évaluations endocriniennes et visuelles [23].
Diagnostic de l'adénome hypophysaire et du syndrome d'hyperprolactinémie
Diagnostic différentiel des masses sellaires/suprasellaires
| Affection | Caractéristiques clés / Points distinctifs | Résultats d'imagerie typiques (IRM) |
|---|---|---|
| Adénome hypophysaire (Micro/Macro) | Symptômes endocriniens (excès/déficit) ou effet de masse (perte de vision, maux de tête, paralysie du nerf crânien). Prend son origine dans la selle turcique, déplace la glande, peut s'étendre aux régions supra/parasellaires. | Micro : Lésion focale <10mm, généralement hypointense en T1, se rehausse moins ou plus tardivement que la glande normale. Macro : Masse sellaire ≥10mm, rehaussement variable (souvent inférieur à la glande), peut présenter une hémorragie ou un kyste, s'étend aux zones supra/parasellaires, peut éroder l'os. |
| Craniopharyngiome | Pic de fréquence bimodal (enfants / personnes âgées). Emplacement suprasellaire fréquent. Maux de tête, perte visuelle, dysfonctionnement endocrinien (déficit en GH, Diabète Insipide fréquent), hydrocéphalie. | Souvent composé de parties kystiques (T1 variable/T2 hyperintense) et solides. La calcification est très courante (mieux visible au scanner). Se rehausse (parties solides, paroi épaisse du kyste). Typiquement à épicentre suprasellaire, peut adhérer aux structures environnantes. |
| Méningiome (Sellaire / Suprasellaire / Sinus Caverneux) | Adultes, plus fréquent chez les femmes. Croissance lente, implanté sur la dure-mère (planum sphénoïdal, tubercule de la selle, diaphragme, paroi du sinus caverneux). Effet de masse (perte de vision, paralysies des NC), maux de tête, convulsions. Rarement une dysfonction endocrinienne significative, sauf si très volumineux. | Souvent isointense en T1/T2, rehaussement intense et homogène après injection de contraste, signe caractéristique de « queue d'implantation durale ». Peut provoquer une hyperostose (épaississement de l'os) ou engainer les vaisseaux. |
| Kyste de la poche de Rathke | Kyste épithélial bénin (vestige de la poche de Rathke), souvent de découverte fortuite. Peut provoquer des maux de tête, des troubles visuels ou un hypopituitarisme s'il est volumineux. Généralement situé en intrasellaire ou en position intra/suprasellaire sur la ligne médiane. | Kyste bien défini ne se rehaussant généralement pas. Le signal varie fortement (T1 hyper/iso/hypo, T2 généralement hyperintense) selon le contenu protéique. Paroi fine et lisse, en principe sans composant solide ni rehaussement (peut présenter un fin liseré). Un nodule intrakystique (protéinacé/hémorragique) est possible (<30%). |
| Hyperplasie hypophysaire | Élargissement du tissu glandulaire normal. Physiologique (grossesse, allaitement, puberté) ou pathologique (réponse à la défaillance de l'organe cible comme dans l'hypothyroïdie primaire, l'insuffisance surrénalienne primaire). | Glande hypertrophiée de manière diffuse (la hauteur peut excéder 10-12 mm), se rehausse le plus souvent de manière intense et homogène. La selle turcique peut s'agrandir symétriquement. La forme de la glande est maintenue (le bord supérieur concave s'efface). Se résorbe si la cause sous-jacente est traitée. |
| Hypophysite (Lymphocytaire, Granulomateuse, à IgG4) | Inflammation de l'hypophyse / de la tige pituitaire. Maux de tête, troubles visuels, hypopituitarisme (souvent d'apparition rapide, touchant plusieurs axes hormonaux), diabète insipide (DI) courant (en particulier lors de l'atteinte de la tige). L'origine lymphocytaire s'observe souvent dans le contexte du post-partum ou d'un processus auto-immun. | Très souvent un élargissement et un épaississement symétrique de la glande / tige, avec un rehaussement intense et diffus. Peut simuler un adénome, mais la tige présente fréquemment un aspect plus infiltrant. Le contexte clinique est crucial (installation rapide d'hypopituitarisme / DI). |
| Métastase | Antécédent de cancer primitif (sein, poumon les plus fréquents). Hypopituitarisme d'installation rapide, le DI est courant (prédilection pour la posthypophyse ou la tige pituitaire). Maux de tête, perte de vision, paralysies de nerfs crâniens possibles. | Lésion se rehaussant, souvent d'aspect infiltratif, impliquant fréquemment la posthypophyse ou la tige pituitaire. Les lésions intracrâniennes peuvent être multiples. La recherche d'une tumeur maligne primitive est primordiale. |
| Anévrisme (Carotide interne intracaverneuse/Suprasellaire) | Peut imiter une masse sellaire, particulièrement si partiellement thrombosé. Peut provoquer des paralysies de NC (III, IV, V, VI à cause de la localisation dans le sinus caverneux), des maux de tête. Risque de rupture (hémorragie sous-arachnoïdienne). | L'IRM montre une disparition du signal d'écoulement (flow void) caractéristique (s'il est perméable), le signal stratifié d'un thrombus (s'il est présent), un rehaussement de la paroi. L'angio-IRM, l'angioscanner ou l'artériographie (DSA) confirment le diagnostic et précisent l'anatomie vasculaire. |
| Chordome / Chondrosarcome | Tumeurs rares, malignes et localement agressives de la base du crâne, issues de reliquats de la chorde dorsale (chordome) ou du cartilage (chondrosarcome). Peuvent s'étendre vers la région sellaire/suprasellaire depuis le clivus ou l'apex pétreux. Entraînent souvent une importante destruction osseuse, paralysies des nerfs crâniens. | Le scanner montre une destruction de l'os. L'IRM montre une masse destructrice localement, prenant le contraste, souvent hyperintense en T2 (spécialement le chordome du fait des cellules physaliphores). Généralement centré sur le clivus ou la jonction pétro-clivale. |
| Germinome (Suprasellaire) | Tumeur germinale rare, principalement chez les enfants ou les jeunes adultes. Triade classique : Diabète Insipide, perte de vision, hypopituitarisme. Peut toucher la région pinéale de manière concomitante (« tumeurs doubles de la ligne médiane »). | Masse suprasellaire avec un rehaussement homogène, impliquant fréquemment le chiasma optique, la tige pituitaire/l'hypothalamus. Les marqueurs tumoraux sériques ou dans le LCR (AFP, β-hCG) peuvent être élevés (bien qu'ils soient souvent normaux en cas de germinome pur). Hautement radiosensible. |
Le diagnostic implique souvent l'Imagerie par Résonance Magnétique (IRM) de l'hypophyse, l'examen de référence pour visualiser les adénomes (microadénomes, macroadénomes, dont les prolactinomes), évaluer leur taille, leur extension, leur relation avec les structures voisines telles que le chiasma optique et les sinus caverneux, et pour aider à les différencier des autres lésions sellaires.
L'approche diagnostique combine l'évaluation clinique, les tests hormonaux et l'imagerie :
- Suspicion clinique : Basée sur les symptômes d'excès hormonal (par ex., traits d'acromégalie, signes cushingoïdes, galactorrhée/aménorrhée), d'effet de masse (changements visuels, maux de tête, paralysie d'un nerf crânien), d'hypopituitarisme (fatigue, hypogonadisme, intolérance au froid, etc.), ou sur une découverte fortuite lors de l'imagerie.
- Bilan hormonal : La mesure des taux de base des hormones de l'hypophyse antérieure et de celles de leurs organes cibles est primordiale pour repérer un excès ou un déficit hormonal. Les examens clés comprennent :
- Prolactine (PRL) : Des niveaux accrus suggèrent une hyperprolactinémie. Des taux très nettement élevés (>200-250 ng/mL ou mcg/L) évoquent très fortement un prolactinome (spécialement en cas de macroadénome). Des taux modérément élevés (25-150 ng/mL) requièrent l'évaluation d'autres causes (médicaments, hypothyroïdie, effet de compression de la tige pituitaire, etc.). Il faut rechercher un « effet crochet » (hook effect) via une dilution de l'échantillon si l'on est en présence d'un macroadénome et que la PRL est anormalement basse/normale [24].
- Hormone de croissance (GH) / Facteur de croissance insulinomimétique de type 1 (IGF-1) : Un taux d'IGF-1 élevé, pondéré selon l'âge et le sexe, est le test de dépistage le plus fiable pour l'acromégalie. Le diagnostic s'affirme par une absence de freination de la GH (< 0,4 ou < 1 ng/mL selon la méthode d'analyse) lors d'une Hyperglycémie Provoquée par Voie Orale (HGPO). Les niveaux basaux de GH s'avèrent moins fiables en raison de leur sécrétion pulsatile.
- ACTH / Cortisol : Le dépistage du syndrome de Cushing repose sur des tests comme la cortisolurie des 24 heures (CLU), le cortisol salivaire nocturne ou encore le test de freinage minute par une faible dose de dexaméthasone (1mg). En cas de positivité, le dosage de l'ACTH plasmatique permet de discerner les origines dépendantes de l'ACTH (origine hypophysaire / ectopique) de celles qui ne le sont pas (surrénalienne). Des tests de freinage fort à la dexaméthasone et/ou des tests de stimulation par la CRH, parfois associés à un cathétérisme des sinus pétreux inférieurs (IPSS), contribuent à affirmer l'origine hypophysaire (Maladie de Cushing) [4].
- TSH / T4 libre/T3 : Une TSH augmentée ou anormalement dans les normes, accompagnée de taux accrus de T4/T3 libres, laisse supposer un rare TSH-ome. Les examens permettent également de dépister une hypothyroïdie périphérique (TSH haute, T4L basse), une origine envisageable de l'hyperprolactinémie secondaire.
- LH / FSH / Testostérone (hommes) / Oestradiol (femmes) : Analysent la fonction des gonades. Des taux diminués témoignent d'un hypogonadisme (d'origine centrale si LH/FSH sont pareillement faibles/dans la norme). La sous-unité alpha est souvent augmentée dans les adénomes gonadotropes.
- Évaluation de l'Hypopituitarisme : Si un effet de masse ou la clinique orientent vers une carence, procéder au dosage à jeun du cortisol et de l'ACTH, de la TSH et de la T4 libre, de la LH/FSH ainsi que de la testostérone/oestradiol (selon le cas), et de l'IGF-1 (pour l'axe somatotrope). Des tests de stimulation dynamiques (par ex., test au Synacthène, test de tolérance à l'insuline – examen de référence mais non dénué de risques, test GHRH-arginine) peuvent être requis pour attester de carences minimes. Vérifier l'existence d'un diabète insipide (DI) en cas de lésion de la posthypophyse (syndrome polyuro-polydipsique, osmolarité sanguine/urinaire).
- Imagerie :
- L'IRM Hypophysaire : Sans et avec injection de Gadolinium, via des séquences fines spécifiques (≤3mm) centrées sur la selle turcique dans les plans coronal et sagittal. Les séquences dynamiques avec contraste sont particulièrement pertinentes pour l'identification des microadénomes, lesquels se manifestent fréquemment par des zones focales au rehaussement retardé comparativement à l'adénohypophyse saine qui capte le produit de contraste rapidement. L'IRM définit avec exactitude les dimensions de la tumeur, ses rapports avec le chiasma optique, la présence d'un envahissement du sinus caverneux (classification de Knosp), et un éventuel déplacement de la tige pituitaire [25].
- Le Scanner (TDM) : Moins performant pour la détection des adénomes à proprement parler, il se révèle plus adapté pour mettre en évidence une érosion osseuse du plancher de la selle turcique ou des clinoïdes, de même que la présence de calcifications (se rencontrant plus volontiers dans les craniopharyngiomes). Il est utile lorsque l'IRM est contre-indiquée.
- Bilan ophtalmologique : Une évaluation réglementaire du champ visuel (souvent par périmétrie automatisée, comme le champ visuel de Humphrey) est incontournable si un adénome est localisé à proximité du chiasma optique ou s'il le comprime (cas des macroadénomes), y compris si le patient ne signale aucun trouble de la vue. L'analyse de l'acuité visuelle et de l'aspect du nerf optique (fond d'œil à la recherche d'une pâleur, d'une atrophie ou d'un œdème papillaire) constitue de la même manière un examen de routine.
- Diagnostic différentiel de l'Hyperprolactinémie : En cas de constatation d'une hausse de la prolactine, il convient d'exclure méthodiquement d'autres étiologies avant de poser le diagnostic de prolactinome, notamment si les concentrations ne sont que modérément accrues (<150 ng/mL) ou si l'imagerie s'avère normale/douteuse. À envisager :
- Des causes physiologiques : La grossesse, la lactation, le stress, le sommeil (contrôler les valeurs matinales), l'activité physique, la stimulation des mamelons, les relations sexuelles.
- Les médicaments : Analyser la liste des traitements actuels (les neuroleptiques - en particulier la rispéridone, les neuroleptiques classiques ; les antidépresseurs - ISRS, tricycliques ; les antiémétiques - métoclopramide, dompéridone ; les traitements antihypertenseurs - vérapamil, méthyldopa ; les opiacés ; les estrogènes). Arrêter le médicament suspect dans la mesure du possible et contrôler la PRL après environ 3 jours (ou en fonction de la demi-vie si elle est plus longue).
- Des pathologies systémiques : L'hypothyroïdie primaire (vérifier TSH/T4L), l'insuffisance rénale chronique (vérifier la créatinine), la cirrhose hépatique sévère, le syndrome des ovaires polykystiques (SOPK), des lésions de la paroi thoracique (zona, cicatrices postopératoires, piercings).
- Syndrome de déconnexion hypothalamo-hypophysaire (Effet tige) : La compression de la tige pituitaire par des processus tumoraux non sécréteurs de prolactine (NFPAs, craniopharyngiomes, méningiomes) ou par des lésions d'ordre inflammatoire bloque l'acheminement de la dopamine de l'hypothalamus vers les cellules lactotropes, ce qui engendre une hyperprolactinémie légère à modérée (généralement <150 ng/mL). L'imagerie est cruciale.
- Macroprolactinémie : L'existence de complexes de prolactine de grande taille, avec une activité biologique moindre ; nécessite des tests de laboratoire précis (précipitation au PEG) ; cliniquement, cela s'avère le plus souvent sans importance.
Les niveaux de prolactine sérique normaux fluctuent légèrement selon le laboratoire et la technique d'analyse employée mais sont habituellement les suivants (intervalles approximatifs) :
Groupe de Patients |
Plage de Prolactine Normale Approximative (ng/mL ou mcg/L) |
(Unités SI Approximatives : mIU/L) |
|---|---|---|
Enfants (pré-pubères) |
Variable, généralement < 15-20 | ~ < 300-400 |
Hommes adultes |
< 15-20 | ~ < 300-400 |
Femmes adultes non enceintes |
< 25-30 | ~ < 500-600 |
Femmes enceintes |
Considérablement élevée, croît tout au long de la grossesse (par ex., jusqu'à 200-400+ ng/mL) | ~ jusqu'à 4000-8000+ mIU/L |
Post-partum (allaitantes) |
Élevée, les valeurs oscillent avec l'allaitement maternel, baissent progressivement si absence d'allaitement | Variable, peut se révéler très importante au commencement |
Le taux de conversion est une approximation (1 ng/mL ≈ 21 mIU/L en se fondant sur les références de l'OMS, bien que cela varie selon le type d'analyse).

Traitement de l'adénome hypophysaire et du syndrome d'hyperprolactinémie
Les objectifs du traitement des adénomes hypophysaires sont de normaliser les niveaux hormonaux (si en excès), de soulager les symptômes d'effet de masse (perte visuelle, maux de tête, paralysies des nerfs crâniens), de préserver ou restaurer la fonction hypophysaire normale, et de prévenir la récidive tumorale. L'approche spécifique dépend fortement du type d'adénome (fonctionnel ou non fonctionnel), de sa taille (micro vs macro), de l'hormone sécrétée, de la présence et de la sévérité de l'effet de masse, de l'âge du patient, de ses projets de grossesse, et des comorbidités [26, 2, 4, 23].
- Prolactinomes (Hyperprolactinémie) :
- Thérapie médicale (Première intention) : Les agonistes dopaminergiques (AD) sont hautement efficaces et constituent le traitement initial de choix pour pratiquement tous les prolactinomes, y compris les grands macroadénomes provoquant des anomalies du champ visuel (à l'exception des cas où la perte de vision est grave ou d'évolution très rapide).
- Cabergoline : Habituellement privilégiée du fait de son efficacité supérieure pour ramener la prolactine à la normale et réduire le volume de la tumeur, de sa demi-vie prolongée permettant une posologie d'une ou deux fois par semaine, et de sa meilleure tolérance en comparaison avec la bromocriptine.
- Bromocriptine : Médicament plus ancien, exigeant une prise tous les jours, plus fréquemment lié à des nausées ou une hypotension orthostatique, bien qu'il jouisse d'un recul important concernant sa sécurité, en particulier lors d'une grossesse.
- Chirurgie (Intervention neurochirurgicale - Chirurgie transsphénoïdale) : Elle est indiquée en premier lieu chez les patients intolérants ou réfractaires aux agonistes dopaminergiques, ceux souffrant d'une apoplexie hypophysaire au sein d'un prolactinome, lors de rares situations impliquant de vastes composantes kystiques ou hémorragiques qui ne réagissent pas aux AD, ou parfois chez des femmes désirant être enceintes et qui ne peuvent supporter ou veulent éviter les AD. Les probabilités de guérison sont supérieures pour les microadénomes (environ 70 à 90 %) en comparaison avec les macroadénomes de type invasif (environ 30 à 50 %).
- Radiothérapie (Radiochirurgie stéréotaxique ou radiothérapie fractionnée) : Destinée à des situations exceptionnelles impliquant des prolactinomes invasifs ou agressifs qui sont réfractaires ou qui récidivent en dépit d'une thérapie médicale et d'une approche chirurgicale optimales. Utilisée avant tout pour freiner l'évolution de la tumeur, l'incidence sur la concentration de prolactine s'avérant lente et la plupart du temps partielle ; le risque d'induire un hypopituitarisme est élevé.
- Thérapie médicale (Première intention) : Les agonistes dopaminergiques (AD) sont hautement efficaces et constituent le traitement initial de choix pour pratiquement tous les prolactinomes, y compris les grands macroadénomes provoquant des anomalies du champ visuel (à l'exception des cas où la perte de vision est grave ou d'évolution très rapide).
- Acromégalie (Adénomes sécrétant de la GH) :
- Chirurgie (En général de première intention) : La chirurgie transsphénoïdale constitue la thérapie initiale dont le but est une résection totale ainsi qu'une rémission biochimique (restauration d'un taux d'IGF-1 conforme aux normes de l'âge et du sexe et un abaissement du taux de GH au cours de l'HGPO). Les chances de succès de cette intervention s'évaluent en fonction du volume du processus tumoral, de son caractère invasif, mais également de l'expérience du praticien.
- Traitement médical : Prescrit dans le cas d'une affection persistant à la suite d'une opération, chez les patients pour qui la chirurgie n'est pas envisageable, ou par moments de manière pré-opératoire dans le but d'optimiser l'efficacité de la chirurgie. Les options de traitement sont :
- Les ligands des récepteurs de la somatostatine (Analogues de la somatostatine) : Les traitements de première génération (octréotide LAR, lanréotide Autogel) rétablissent les valeurs d'IGF-1/GH chez 50 à 60 % des patients et sont en mesure de réduire la masse tumorale. Ceux de deuxième génération (pasiréotide LAR) peuvent s'avérer pertinents lorsque les molécules de la génération précédente s'avèrent inefficaces, cependant, ils s'accompagnent d'un risque accru de développement d'hyperglycémie.
- Antagoniste de l'hormone de croissance (Pegvisomant) : Très performant pour restaurer un niveau d'IGF-1 adéquat (environ 80 à 90 %) en entravant l'activité de la GH au niveau de son récepteur ; il n'engendre pas de réduction du volume tumoral et implique une surveillance des fonctions hépatiques et de la taille de la tumeur.
- Agonistes Dopaminergiques (Cabergoline) : D'une efficacité moindre, il ne rétablit les valeurs de l'IGF-1 que chez environ 10 à 30 % des personnes, il est principalement administré en tant que thérapie complémentaire, en particulier lorsque la tumeur sécrète également de la prolactine.
- Radiothérapie (Stéréotaxique ou Fractionnée) : Performante pour stopper la croissance tumorale et pour diminuer de façon progressive les valeurs de GH/IGF-1 chez les sujets présentant un reliquat tumoral ou une récurrence, insensibles à la thérapie médicale. Probabilité importante de développer un hypopituitarisme à retardement.
- Maladie de Cushing (Adénomes sécrétant de l'ACTH) :
- Chirurgie (De première intention) : L'adénomectomie sélective par voie transsphénoïdale est l'approche thérapeutique de prédilection, avec pour objectif d'atteindre une rémission sur le plan biochimique tout en préservant l'intégrité fonctionnelle de la glande pituitaire. Les probabilités de rémission se situent aux alentours de 70 à 90 % en cas de microadénomes lorsque l'intervention est menée par des établissements hautement qualifiés [27].
- Traitement médical : Employé préalablement à l'opération dans le but de maîtriser un hypercorticisme intense, en phase postopératoire si l'affection persiste ou s'il y a rechute en attendant l'efficacité de la radiothérapie, ou comme approche principale lorsque la chirurgie n'a pas abouti ou s'avère proscrite. Les médicaments interagissent avec le processus de stéroïdogenèse au sein des glandes surrénales (kétoconazole, métyrapone, étomidate, osilodrostat), la libération d'ACTH par la glande pituitaire (pasiréotide, cabergoline), ou procèdent au blocage des récepteurs aux glucocorticoïdes (mifépristone).
- Radiothérapie (Stéréotaxique ou Fractionnée) : Performante dans les situations de maladie persistante ou de rechute suite à une intervention chirurgicale, néanmoins la rémission biochimique est différée (plusieurs mois à quelques années).
- Surrénalectomie bilatérale : Permet d'éradiquer de façon certaine l'hypercorticisme lorsque les traitements focalisés sur l'hypophyse s'avèrent inefficaces, cependant, elle engendre une carence surrénalienne irréversible, rendant indispensable un traitement compensatoire à vie par glucocorticoïdes/minéralocorticoïdes, et expose à l'éventualité du développement du syndrome de Nelson (développement évolutif et hyperpigmentation engendrés par la tumeur de l'hypophyse qui synthétise de l'ACTH).
- Adénome thyréotrope (TSH-ome, rare) :
- Chirurgie (De première intention) : L'exérèse transsphénoïdale représente le traitement de choix.
- Thérapie médicale : Les analogues de la somatostatine (octréotide, lanréotide) démontrent fréquemment leur efficacité dans la réduction de la libération de TSH ainsi que dans la diminution de la taille de la tumeur préalablement à la chirurgie ou pour un reliquat tumoral. Les médicaments antithyroïdiens (méthimazole, PTU) traitent la symptomatologie de l'hyperthyroïdie, sans pour autant traiter l'adénome.
- Radiothérapie : Pour les reliquats de tumeurs ou les récidives insensibles aux autres formes de traitement.
- Adénomes hypophysaires non fonctionnels (NFPAs) : Se référer à la rubrique ci-après.
Les choix de prise en charge nécessitent d'être personnalisés et impliquent souvent la collaboration d'une équipe pluridisciplinaire regroupant des endocrinologues, neurochirurgiens, neuroradiologues, ophtalmologistes et, le cas échéant, des spécialistes en radio-oncologie.
Traitement des adénomes hypophysaires non fonctionnels (NFPAs)
Les NFPAs ne provoquant généralement pas de syndromes associés à une surproduction hormonale, une intervention est recommandée avant tout s'ils sont à l'origine d'un effet de masse (perte de la vision, atteintes des nerfs crâniens, céphalées cliniquement prononcées et imputables au processus tumoral) ou s'ils entraînent un hypopituitarisme, ou bien encore s'ils présentent une progression notable lors d'examens d'imagerie séquentiels [22, 23].
- NFPAs de découverte fortuite et asymptomatiques :
- Microadénomes (<10mm) : Ils font majoritairement l'objet d'une prise en charge non invasive avec un suivi périodique par IRM (par exemple, IRM à un an, par la suite tous les 1 à 2 ans, puis de façon plus espacée si la situation est stabilisée) associée à une évaluation endocrinienne de référence (recherche d'un hypopituitarisme et d'une hyperprolactinémie causée par une compression de la tige). La majeure partie (environ 90 à 95 %) n'évolue pas de manière notable [8].
- Macroadénomes (≥10mm) dépourvus d'effet de masse (Absence de troubles visuels, activité hypophysaire intacte, ne comprimant pas le chiasma) : L'approche est personnalisée. Les possibilités englobent une surveillance attentive via des IRM (par exemple à un intervalle de 6 mois, puis de façon annuelle si la tumeur est stabilisée) et des examens endocriniens/visuels périodiques, ou la possibilité d'une intervention chirurgicale, particulièrement si la tumeur est volumineuse ou se trouve à très grande proximité du chiasma optique, compte tenu du risque d'une expansion future ou de survenue de complications. L'intervention est prescrite en cas d'expansion tumorale attestée, de l'apparition de manifestations (altération de la vue, hypopituitarisme) ou si la tumeur entre en contact avec/comprime le chiasma optique [23].
- NFPAs symptomatiques (provoquant un effet de masse ou un hypopituitarisme) :
- Chirurgie (Première ligne) : L'intervention par la voie transsphénoïdale constitue la procédure de référence. Les objectifs fondamentaux résident dans la levée de la compression sur le système visuel afin de restaurer ou de sauvegarder les capacités visuelles et l'ablation la plus complète possible et sécurisée de la tumeur pour pallier l'effet de masse et possiblement favoriser la reprise de l'activité de l'hypophyse (une éventualité plus incertaine en présence de troubles établis depuis longtemps). L'utilisation de techniques endoscopiques par les fosses nasales est devenue courante de nos jours. L'exérèse totale s'avère souvent complexe dans le cadre de tumeurs de grand volume ou de caractère invasif (par exemple, s'infiltrant dans le sinus caverneux).
- Radiothérapie (Radiochirurgie Stéréotaxique - SRS ou Radiothérapie Fractionnée) : Orientée avant tout comme modalité d'appoint en présence d'un reliquat tumoral notable suite à l'opération (particulièrement si celui-ci montre des signes d'évolution ou si le volume restant est important), ou en présence de tumeurs en récidive. La radiothérapie en première intention est très exceptionnellement mise en œuvre, sauf en cas d'impossibilité de pratiquer une intervention chirurgicale. La radiothérapie garantit des taux remarquables de stabilisation des tumeurs sur le long terme (>90 %) mais ne pallie pas de manière instantanée l'effet de masse et implique un risque retardé substantiel (~30-60 % sur une décennie) de survenue d'un hypopituitarisme secondaire à l'irradiation [28].
- Thérapie médicale : Actuellement, aucune intervention pharmacologique n'a démontré son aptitude à diminuer systématiquement la taille des NFPAs ni à entraver leur développement. Bien que les agonistes dopaminergiques et les analogues de la somatostatine aient fait l'objet d'études, ils se révèlent le plus souvent sans efficacité pour les NFPAs d'origine gonadotrope classiques.
- Prise en charge de l'hypopituitarisme : Toute carence hormonale mise en évidence préalablement ou à la suite d'un traitement exige la mise en place d'un traitement de substitution adéquat et définitif (par ex., hydrocortisone en cas d'insuffisance surrénalienne, avec nécessité d'ajuster les doses en situation de stress ; lévothyroxine pour parer à une hypothyroïdie d'origine centrale ; testostérone/œstrogènes associés ou non à la progestérone lors d'un hypogonadisme ; de façon potentielle, un remplacement par GH chez l'adulte face à une déficience attestée et ayant une forte incidence sur le confort de vie ou les indicateurs métaboliques ; administration de DDAVP si présence d'un diabète insipide).
- Surveillance au long cours : Elle s'avère primordiale à l'issue des traitements (chirurgie associée ou non à la radiothérapie) pour la totalité des macroadénomes. Cette surveillance implique de contrôler l'absence de récidive ou la progression tumorale (IRM sériées, dont la périodicité est conditionnée par l'existence d'un reliquat et de la méthode thérapeutique), l'examen régulier des fonctions hormonales (bilan annuel à la recherche d'un hypopituitarisme, particulièrement post-radiothérapie), et l'évaluation de la fonction visuelle (examen du champ visuel lors d'antécédents de compression). L'incidence des récurrences après une chirurgie isolée peut être notable avec les années (10 à 50 % en fonction de l'intervalle de suivi et de l'importance du reliquat), ce qui impose de s'interroger sur l'opportunité d'une radiothérapie d'appoint en présence de résidus tumoraux préoccupants.
![]() Attention ! Les adénomes de l'hypophyse requièrent une évaluation diagnostique et un suivi spécialisés dispensés par une équipe pluridisciplinaire. L'apparition de symptômes tels qu'une altération de la vision, des céphalées opiniâtres, des troubles endocriniens (anomalies du cycle menstruel, écoulement lacté, modifications corporelles non justifiées, troubles de la fonction sexuelle), ou un épuisement inhabituel, appelle à une consultation médicale, laquelle pourra orienter vers une prise en charge conjointe par un endocrinologue et un neurochirurgien.
Attention ! Les adénomes de l'hypophyse requièrent une évaluation diagnostique et un suivi spécialisés dispensés par une équipe pluridisciplinaire. L'apparition de symptômes tels qu'une altération de la vision, des céphalées opiniâtres, des troubles endocriniens (anomalies du cycle menstruel, écoulement lacté, modifications corporelles non justifiées, troubles de la fonction sexuelle), ou un épuisement inhabituel, appelle à une consultation médicale, laquelle pourra orienter vers une prise en charge conjointe par un endocrinologue et un neurochirurgien.
Références
- Melmed S. Pituitary-Tumor Endocrinopathies. N Engl J Med. 2020 Mar 5;382(10):937-950. doi: 10.1056/NEJMra1810772
- Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011 Feb;96(2):273-88. doi: 10.1210/jc.2010-1692
- Katznelson L, Laws ER Jr, Melmed S, et al; Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Nov;99(11):3933-51. doi: 10.1210/jc.2014-2700
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008 May;93(5):1526-40. doi: 10.1210/jc.2008-0125
- Beck-Peccoz P, Persani L. Thyrotropin-secreting pituitary adenomas. N Engl J Med. 1996;334(11):712-713. doi: 10.1056/NEJM199603143341107
- Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2011 Jun;14(2):111-8. doi: 10.1007/s11102-011-0300-1
- Molitch ME. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 1997 Mar;26(1):1-17. doi: 10.1016/s0889-8529(05)70239-7
- Fernández-Balsells MM, Murad MH, Barwise A, et al. Clinical management of nonfunctioning pituitary adenomas (NFPAs): a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011 Apr;96(4):905-15. doi: 10.1210/jc.2010-1921
- Chapter on Pituitary Imaging. In: Osborn AG, Salzman KL, Jhaveri MD, et al. Osborn's Brain. 2nd ed. Elsevier; 2018.
- Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. 2002 Feb;2(2):83-94. doi: 10.1038/nrc760
- Vierimaa O, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006 May 26;312(5777):1228-30. doi: 10.1126/science.1126100
- Reincke M, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. 2015 Jan;47(1):31-8. doi: 10.1038/ng.3166
- Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936 Mar;12(2):205-16.1.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004 Aug 1;101(3):613-9. doi: 10.1002/cncr.20412
- Hall WA, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994 May 15;120(10):817-20. doi: 10.7326/0003-4819-120-10-199405150-00001
- Ciccarelli A, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Varese, Italy. J Clin Endocrinol Metab. 2001 Feb;86(2):720-5. doi: 10.1210/jcem.86.2.7204
- Fernandez A, et al. Prevalence of pituitary adenomas: a community-based study in Olmsted County, Minnesota, 1993-1997. Arch Intern Med. 2000 Jan 10;160(1):78-81. doi: 10.1001/archinte.160.1.78
- Daly AF, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006 Dec;91(12):4756-69. doi: 10.1210/jc.2006-1668
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009 Mar;119(3):429-38. doi: 10.1172/JCI38090
- Nieman LK. Cushing's syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015 Oct;173(4):M33-8. doi: 10.1530/EJE-15-0464
- Elster AD, et al. Pituitary gland: MR imaging of physiologic hypertrophy in adolescence. Radiology. 1990;174(3 Pt 1):681-5. doi: 10.1148/radiology.174.3.2305044
- Molitch ME. Nonfunctioning pituitary adenomas. Pituitary. 2005;8(3-4):213-20. doi: 10.1007/s11102-006-8512-6
- Freda PU, Beckers AM, Katznelson L, et al; Endocrine Society. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011 Apr;96(4):894-904. doi: 10.1210/jc.2010-1048
- Frieze TW, et al. Hook effect in prolactinomas: case report and review of literature. Endocr Pract. 2006;12(3):296-303. doi: 10.4158/EP.12.3.296
- Bonneville F, et al. Radiology of pituitary adenomas: an update. Radiol Clin North Am. 2012 Nov;50(6):995-1013. doi: 10.1016/j.rcl.2012.08.005
- Melmed S, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509-17. doi: 10.1210/jc.2008-2421
- Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818
- Sheehan JP, et al. Radiosurgery for nonfunctioning pituitary adenomas: a systematic review. J Neurosurg. 2013 Aug;119(2):446-56. doi: 10.3171/2013.4.JNS121833
Voir aussi
- Anatomie du système nerveux
- Infection du système nerveux central :
- Abcès cérébral (lobaire, cérébelleux)
- Granulome éosinophile, histiocytose langerhansienne (LCH), signe de Hennebert
- Abcès épidural cérébral
- Complications intracrâniennes de la sinusite
- Complications intracrâniennes d'origine otogène
- Complications ophtalmiques de la sinusite
- Méningite bactérienne otogène
- Abcès sous-dural cérébral
- Thrombophlébite suppurée du sinus sigmoïde
- Kyste colloïde du troisième ventricule
- Arachnoïdite adhésive cérébrale et spinale
- Dégénérescence cortico-basale (atrophie cérébrale focalisée)
- Encéphalopathie
- Maux de tête, migraine
- Lésion cérébrale traumatique (commotion, contusion, hémorragie cérébrale, lésions axonales diffuses)
- Hypertension intracrânienne et hydrocéphalie
- Maladie de Parkinson
- Microadénome hypophysaire, macroadénome et adénomes non fonctionnels (NFPAs), syndrome d'hyperprolactinémie
- Fuite spontanée de liquide céphalo-rachidien (rhinorrhée de LCR)