Hypophysen-Mikroadenom, Makroadenom und inaktive Adenome (NFPAs), Hyperprolaktinämie-Syndrom
- Hypophysenadenom (Mikroadenom, Makroadenom), Hyperprolaktinämie-Syndrom Überblick
- Hypophysen-Mikroadenome:
- Hypophysen-Makroadenome:
- Inaktive Hypophysenadenome (NFPAs)
- Hypophysenadenom und Hyperprolaktinämie-Syndrom Diagnose
- Hypophysenadenom und Hyperprolaktinämie-Syndrom Behandlung
- Behandlung inaktiver Hypophysenadenome (NFPAs)
- Referenzen
Hypophysenadenom (Mikroadenom, Makroadenom), Hyperprolaktinämie-Syndrom Überblick
Hypophysenadenome sind typischerweise gutartige (nicht krebsartige) Tumore, die von den hormonproduzierenden Zellen des Hypophysenvorderlappens (Adenohypophyse) ausgehen, der sich an der Schädelbasis in einer knöchernen Struktur, der Sella turcica, befindet [1]. Diese Adenome werden anhand ihrer Größe (Mikroadenom gegenüber Makroadenom) und ihrer hormonellen Aktivität (aktiv gegenüber inaktiv) klassifiziert.
Das Hyperprolaktinämie-Syndrom bezieht sich auf die klinischen Folgen von anhaltend erhöhten Spiegeln von Prolaktin, einem Hormon, das normalerweise an der Laktation beteiligt ist. Prolaktinome (Prolaktin-sezernierende Hypophysenadenome) sind die häufigste Art von aktiven Hypophysenadenomen und eine häufige Ursache für Hyperprolaktinämie [2]. Bei Frauen umfassen die Symptome typischerweise Galaktorrhoe (unangemessene Milchproduktion), Menstruationsstörungen (Oligomenorrhoe oder Amenorrhoe) und Unfruchtbarkeit. Bei Männern führt Hyperprolaktinämie oft zu verminderter Libido, erektiler Dysfunktion, Unfruchtbarkeit aufgrund von Oligospermie, Gynäkomastie (Brustvergrößerung) und seltener zu Galaktorrhoe. Erhöhtes Prolaktin hemmt die Sekretion des Gonadotropin-Releasing-Hormons (GnRH) aus dem Hypothalamus, was zu sekundärem Hypogonadismus führt.

Symptome im Zusammenhang mit Hypophysenadenomen entstehen entweder durch überschüssige Hormonsekretion (bei aktiven Adenomen) oder durch die physische Größe des Tumors und den Druck auf umliegende Strukturen (Masseneffekt), insbesondere bei Makroadenomen. Hormonüberschuss-Syndrome umfassen:
Hormonüberschuss |
Resultierende Erkrankung & Hauptgesundheitsprobleme |
|---|---|
| Prolaktin (PRL) |
Hyperprolaktinämie: - Frauen: Menstruationsstörungen (Oligomenorrhoe, Amenorrhoe), Galaktorrhoe, Unfruchtbarkeit, verminderte Knochendichte. - Männer: Verminderte Libido, erektile Dysfunktion, Unfruchtbarkeit, Gynäkomastie (seltener), verminderte Knochendichte. [2] |
| Wachstumshormon (GH) |
Akromegalie (Erwachsene) / Gigantismus (Kinder): - Vergrößerung von Händen, Füßen, Gesichtszügen (Kiefer, Stirn). - Weichteilschwellung, übermäßiges Schwitzen. - Arthrose, Karpaltunnelsyndrom. - Schlafapnoe. - Bluthochdruck, Kardiomyopathie. - Diabetes mellitus oder gestörte Glukosetoleranz. - Erhöhtes Risiko für Darmpolypen/-krebs. [3] |
| Adrenokortikotropes Hormon (ACTH) |
Morbus Cushing (Hypophysenabhängiges Cushing-Syndrom): - Stammfettsucht (Rumpffett, Vollmondgesicht, Stiernacken), dünne Haut, leichte Blutergüsse, Striae rubrae. - Muskelschwäche (proximale Myopathie), Osteoporose. - Stimmungsschwankungen (Angst, Depression, Psychose). - Bluthochdruck, Diabetes mellitus. - Erhöhte Anfälligkeit für Infektionen. - Menstruationsstörungen (Frauen). [4] |
| Thyreoideastimulierendes Hormon (TSH) |
TSH-om (selten): - Sekundäre Hyperthyreose: Zu den Symptomen gehören Hitzeintoleranz, Schwitzen, Tachykardie/Herzklopfen (Arrhythmien wie Vorhofflimmern), Tremor, Angst/Reizbarkeit, unbeabsichtigter Gewichtsverlust trotz erhöhtem Appetit, Kropf. Gekennzeichnet durch unangemessen normales oder erhöhtes TSH mit erhöhten freien T4/T3-Spiegeln. [5] |
| Gonadotropine (LH, FSH) |
Gonadotropes Adenom (oft klinisch inaktiv): - Sezernieren gewöhnlich inaktive oder intakte Hormonfragmente (z.B. Alpha-Untereinheit, Beta-FSH, seltener intaktes FSH/LH). - Kann selten ovarielle Überstimulation (Frauen) oder Hodenvergrößerung/Pubertas praecox (Männer) verursachen. - Manifestiert sich meistens durch Masseneffekt oder wird zufällig entdeckt. [6] |
Inaktive Hypophysenadenome (NFPAs) sezernieren keine klinisch signifikanten Mengen aktiver Hormone, obwohl viele Hormon-Untereinheiten produzieren, die durch spezielle Tests nachweisbar sind. Sie verursachen typischerweise Symptome durch den Masseneffekt, wenn sie zu Makroadenomen werden, indem sie benachbarte Strukturen wie das Chiasma opticum (was zu Gesichtsfeldausfällen, klassischerweise bitemporaler Hemianopsie, führt), Hirnnerven im Sinus cavernosus (verursacht Doppelbilder) oder normales Hypophysengewebe (verursacht Hypopituitarismus - Mangel an einem oder mehreren Hypophysenhormonen) komprimieren. Kopfschmerzen sind ebenfalls häufig aufgrund der Dehnung der Dura oder erhöhtem sellären Druck.
Hypophysen-Mikroadenome
Beschreibung der Hypophysen-Mikroadenome
Ein Hypophysen-Mikroadenom ist definiert als ein Hypophysenadenom mit einem maximalen Durchmesser von weniger als 10 Millimetern (weniger als 1 cm) [7]. Diese Tumore sind auf die Sella turcica beschränkt und verursachen typischerweise keine Masseneffektsymptome wie Sehverlust. Mikroadenome können entweder aktiv (hormonsezernierend) oder inaktiv sein. Aktive Mikroadenome verursachen Symptome, die mit dem spezifischen Hormon zusammenhängen, das sie überproduzieren (z.B. Prolaktinom verursacht Hyperprolaktinämie, ACTH-sezernierend verursacht Morbus Cushing, GH-sezernierend verursacht Akromegalie).
Viele Mikroadenome sind jedoch klinisch inaktiv und werden zufällig während einer Bildgebung des Gehirns (Magnetresonanztomographie - MRT oder CT) entdeckt, die aus anderen Gründen durchgeführt wird (z.B. Kopfschmerzen, Schwindel, Trauma). Diese werden oft als Hypophysen-"Inzidentalome" bezeichnet. Während die große Mehrheit gutartige Befunde sind, die nur überwacht werden müssen, können selbst kleine inzidentelle Mikroadenome manchmal subtile hormonelle Ungleichgewichte (subklinischer Cushing, milde Hyperprolaktinämie) verursachen oder, selten (< 10 % über mehrere Jahre), im Laufe der Zeit wachsen [8].
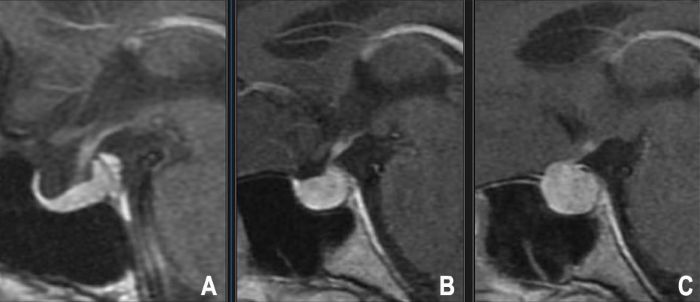
Die MRT mit Gadolinium-Kontrastmittel, insbesondere unter Verwendung dynamischer kontrastverstärkter Sequenzen und dünner Schichten durch die Hypophysengrube, ist die Bildgebungsmodalität der Wahl für den Nachweis von Mikroadenomen. Sie erscheinen oft als kleine, fokale Läsionen, die das Kontrastmittel weniger stark oder später als das umgebende normale Hypophysengewebe aufnehmen (typischerweise hypointens in frühen postkontrast T1-Bildern) [9].
Klinisch signifikante Mikroadenome sind am häufigsten Prolaktinome, gefolgt von ACTH-sezernierenden Adenomen, die Morbus Cushing verursachen, und GH-sezernierenden Adenomen, die Akromegalie verursachen. Inaktive Mikroadenome sind immunhistochemisch oft gonadotrope Adenome, weisen aber keinen klinischen Hormonüberschuss auf. Zur Differentialdiagnose in der Bildgebung gehören Rathke-Zysten, Hypophysenhyperplasie, Metastasen oder entzündliche Prozesse (Hypophysitis), obwohl diese oft unterschiedliche Merkmale oder klinische Kontexte aufweisen.
Pathophysiologie der Hypophysen-Mikroadenome
Hypophysenadenome, einschließlich Mikroadenome, gelten im Allgemeinen als monoklonale Neoplasien, was bedeutet, dass sie aus der klonalen Expansion einer einzelnen mutierten Zelle des Hypophysenvorderlappens entstehen [10]. Die spezifischen initiierenden Mutationen und nachfolgenden genetischen/epigenetischen Ereignisse, die zur Adenombildung führen, sind komplex und vielfältig, aber mehrere Faktoren und Signalwege sind beteiligt.
Spezifische genetische Keimbahnsyndrome prädisponieren Individuen für Hypophysenadenome: Multiple endokrine Neoplasie Typ 1 (MEN1), verursacht durch Mutationen im MEN1-Tumorsuppressorgen, betrifft häufig Prolaktinome oder GH-sezernierende Adenome. Das McCune-Albright-Syndrom, bedingt durch mosaikartige aktivierende Mutationen im GNAS-Gen, das für die Gs-alpha-Proteinuntereinheit kodiert, führt oft zu GH-sezernierenden Adenomen (Somatotropinomen). Der Carney-Komplex (PRKAR1A-Mutationen) kann Hypophysenadenome umfassen (typischerweise GH-sezernierend). Mutationen im AIP-Gen (Aryl-Hydrocarbon-Rezeptor-Interacting-Protein) prädisponieren für familiäre isolierte Hypophysenadenome (FIPA), insbesondere GH-sezernierende oder Prolaktin-sezernierende Tumore, die sich oft im jungen Alter manifestieren und größer/aggressiver sind [11].
Sporadische (nicht familiäre) Adenome weisen ebenfalls somatische genetische und epigenetische Veränderungen auf. Aktivierende somatische Mutationen in GNAS finden sich in etwa 40 Prozent der sporadischen GH-sezernierenden Adenome. Mutationen in USP8 sind bei ACTH-sezernierenden Adenomen häufig [12]. Andere somatische Mutationen und epigenetische Veränderungen, die Zellzyklus-Regulatoren (z.B. Funktionsverlust in CDKN1B/p27, Veränderungen in p53, RB-Wegen), Wachstumsfaktor-Wege (z.B. Überexpression von FGFR4) und hypophysenspezifische Transkriptionsfaktoren beeinflussen, tragen zur Tumorentstehung und Hormonproduktion bei. Ein Heterozygotieverlust an bestimmten chromosomalen Orten (z.B. 11q13, wo sich MEN1 befindet) wird ebenfalls beobachtet.
Prolaktinome, der häufigste Typ, entstehen aus laktotropen Zellen. Kortikotropinome, die Morbus Cushing verursachen, entstehen aus kortikotropen Zellen. Somatotropinome, die Akromegalie verursachen, entstehen aus somatotropen Zellen. Gonadotropinome entstehen gewöhnlich aus gonadotropen Zellen und sezernieren oft inaktive Hormonuntereinheiten (Alpha-Untereinheit, Beta-FSH, Beta-LH). Thyreotropinome, die Hyperthyreose verursachen, entstehen aus thyreotropen Zellen und sind selten.
Die meisten klinisch inaktiven Adenome sind immunpositiv für Gonadotropin-Untereinheiten (FSH, LH, Alpha-Untereinheit), verursachen aber keine klinischen Syndrome des Hormonüberschusses. Einige können "Nullzell"-Adenome ohne spezifische Hormonfärbung oder stumme kortikotrope/somatotrope Adenome sein.
Epidemiologie der Hypophysen-Mikroadenome
Häufigkeit von Hypophysen-Mikroadenomen
Hypophysen-Mikroadenome sind überraschend häufige Zufallsbefunde. Autopsiestudien haben mikroskopische oder kleine Adenome (oft < 3 mm) bei bis zu 27 Prozent der Personen ohne bekannte Hypophysenerkrankung berichtet [13, 14]. Hochauflösende Bildgebungsstudien (MRT) entdecken Hypophysen-Inzidentalome (von denen die meisten Mikroadenome < 10 mm sind) bei etwa 10-20 Prozent der Allgemeinbevölkerung, die aus nicht zusammenhängenden Gründen Gehirnscans unterzogen werden [15].
Die überwiegende Mehrheit dieser zufällig entdeckten Mikroadenome ist inaktiv und bleibt stabil oder wächst sehr langsam, ohne jemals klinische Symptome zu verursachen. Aktive Mikroadenome sind viel seltener, stellen aber die übliche Ursache für Hypophysenhormon-Überschusssyndrome wie Hyperprolaktinämie (wobei das Prolaktinom der am häufigsten klinisch diagnostizierte Typ ist, geschätzte Prävalenz ~10-50 pro 100.000 Einwohner [16]), Morbus Cushing und Akromegalie dar.
Hypophysen-Mikroadenome können in jedem Alter auftreten, werden aber am häufigsten bei jungen bis mittelalten Erwachsenen (20er-50er Jahre) diagnostiziert. Es gibt ein Überwiegen der Frauen bei Prolaktinomen (Verhältnis F:M ~10:1) und Morbus Cushing (~3-5:1), während GH-sezernierende Adenome eine in etwa gleiche Geschlechterverteilung aufweisen.
Vor der weitverbreiteten Anwendung der MRT war die durch Bluttests nachgewiesene Hyperprolaktinämie ein wichtiger Hinweis auf zugrunde liegende, oft ungesehene, Prolaktin-sezernierende Mikroadenome. Die MRT ermöglicht nun die direkte Visualisierung.
Die hohe Prävalenz von Mikroadenomen, die zufällig gefunden werden, im Vergleich zur geringeren Prävalenz von klinisch signifikanten Hypophysenerkrankungen oder Makroadenomen deutet darauf hin, dass die meisten Mikroadenome nicht zu großen oder symptomatischen Tumoren voranschreiten.
Hypophysen-Makroadenome
Beschreibung der Hypophysen-Makroadenome
Ein Hypophysen-Makroadenom ist definiert als ein Hypophysenadenom mit einem maximalen Durchmesser von 10 Millimetern (1 cm) oder mehr [3]. Im Gegensatz zu Mikroadenomen breiten sich Makroadenome häufig über die Grenzen der Sella turcica hinaus aus und komprimieren potenziell umliegende neuronale und vaskuläre Strukturen.
Sie können entweder aktiv (hormonsezernierend) oder inaktiv sein. Inaktive Hypophysenadenome (NFPAs) sind die häufigste Art von Makroadenom, die klinisch diagnostiziert wird [6]. Aktive Makroadenome umfassen große Prolaktinome, GH-sezernierende Adenome, die Akromegalie verursachen, und seltener große ACTH-sezernierende Adenome (oft assoziiert mit dem Nelson-Syndrom nach Adrenalektomie) oder TSH-sezernierende Adenome.
Makroadenome präsentieren sich aufgrund ihrer Größe oft mit Symptomen, die mit dem Masseneffekt zusammenhängen:
- Sehstörungen: Die Kompression des Chiasma opticum (Sehnervenkreuzung), das sich superior der Hypophyse befindet, ist bei suprasellärer Ausdehnung häufig und verursacht klassischerweise eine bitemporale Hemianopsie (Verlust des peripheren Sehens in beiden äußeren temporalen Gesichtsfeldern). Die Kompression der Sehnerven oder des Tractus opticus kann andere Muster des Sehverlusts oder eine verringerte Sehschärfe verursachen.
- Hirnnervenlähmungen: Die laterale Ausdehnung in den Sinus cavernosus kann die Hirnnerven III (N. oculomotorius), IV (N. trochlearis), V (N. trigeminus - Augen- und Oberkieferäste V1 und V2) und VI (N. abducens) komprimieren, was zu Doppelbildern (Diplopie), hängendem Augenlid (Ptosis), Pupillenveränderungen oder Gesichtstaubheit/-schmerzen führt. HN III und VI sind am häufigsten klinisch betroffen.
- Kopfschmerzen: Oft unspezifisch, möglicherweise aufgrund der Dehnung der Dura mater, die die Sella bedeckt (Diaphragma sellae), oder eines erhöhten intrasellären Drucks.
- Hypopituitarismus: Kompression und Zerstörung des normalen Hypophysengewebes können zu einem Mangel an einem oder mehreren Hormonen des Hypophysenvorderlappens führen (GH-Mangel ist oft der früheste, gefolgt von Gonadotropinen LH/FSH, was zu Hypogonadismus führt, dann TSH, was zu sekundärer Hypothyreose führt, und schließlich ACTH, was zu sekundärer Nebennierenrindeninsuffizienz führt) oder, seltener, zu einer Dysfunktion des Hypophysenhinterlappens (Mangel an antidiuretischem Hormon - ADH, der einen zentralen Diabetes insipidus verursacht, häufiger gesehen bei Stielkompression oder Läsionen, die posterior entstehen).
- Hypophysenapoplex: Ein akuter medizinischer Notfall, der durch plötzliche Blutung oder Infarkt innerhalb eines Hypophysenadenoms verursacht wird (bei Makroadenomen viel häufiger als bei Mikroadenomen). Präsentiert sich mit plötzlichen, starken Kopfschmerzen, schnellem Sehverlust, Hirnnervenlähmungen, verändertem Bewusstsein und oft akutem Hypopituitarismus (erfordert dringenden Steroidersatz).
Aktive Makroadenome verursachen auch Symptome aufgrund der Hormonüberproduktion, die oft schwerer sind als bei Mikroadenomen, da die Tumorlast größer ist, wie zuvor beschrieben.
Pathophysiologie der Hypophysen-Makroadenome
Die zugrunde liegende Pathophysiologie beinhaltet die klonale Expansion mutierter adenohypophysärer Zellen, ähnlich wie bei Mikroadenomen. Die Faktoren, die die Progression vom Mikroadenom zum Makroadenom oder die de novo-Entwicklung von Makroadenomen vorantreiben, sind nicht vollständig geklärt, beinhalten aber wahrscheinlich die Akkumulation zusätzlicher genetischer (z.B. Mutationen in Tumorsuppressorgenen wie p53, chromosomale Instabilität) oder epigenetischer Veränderungen, die eine erhöhte Proliferation, verminderte Apoptose, Angiogenese und manchmal Invasivität fördern [10].
Makroadenome zeigen oft einen Verlust normaler Rückkopplungsmechanismen, die die Hormonsekretion kontrollieren (besonders bei aktiven Tumoren). Ihr Wachstum führt zur Kompression und Zerstörung benachbarter normaler Hypophysenzellen, was zum Hypopituitarismus führt. Die Extension nach oben komprimiert das Chiasma opticum und potenziell den Hypothalamus (was zu DI oder Fehlregulation von Appetit/Temperatur führt). Die laterale Ausdehnung invadiert den Sinus cavernosus, was potenziell die Arteria carotis interna und die Hirnnerven III, IV, V1, V2 und VI umhüllt. Die inferiore Ausdehnung kann den Boden der Sella turcica in die Keilbeinhöhle erodieren und manchmal eine Liquor-Rhinorrhoe verursachen.
Die genetische Basis umfasst Faktoren, die für Mikroadenome erwähnt wurden, einschließlich Keimbahnsyndrome (MEN1, GNAS, AIP, Carney-Komplex) und somatische Mutationen (GNAS, USP8). Zusätzliche Veränderungen, die mit größeren oder aggressiveren Tumoren assoziiert sind, könnten Mutationen in Zellzyklus-Regulatoren (p53, RB), Wachstumsfaktorrezeptoren (FGFR4) oder Adhäsionsmolekülen umfassen. Während sie histologisch gutartig sind (niedrige Mitoserate, gewöhnlich keine zytologische Atypie), zeigen einige Makroadenome invasive Wachstumsmuster in umliegende Strukturen (Dura, Knochen, Sinus cavernosus), was eine vollständige chirurgische Resektion schwierig macht. Echte primäre Hypophysenkarzinome, definiert durch das Vorhandensein kraniospinaler oder systemischer Metastasen, sind äußerst selten.
Assoziierte klinische Syndrome wie MEN1, McCune-Albright und Carney-Komplex können Makroadenome umfassen, oft GH-sezernierende oder Prolaktin-sezernierende Typen.
Epidemiologie der Hypophysen-Makroadenome
Häufigkeit von Hypophysen-Makroadenomen
Während zufällige Mikroadenome sehr häufig sind, sind klinisch signifikante Hypophysentumoren, die eine Beurteilung und potenzielle Behandlung erfordern, seltener. Die Gesamtprävalenz klinisch manifester Hypophysenadenome wird auf etwa 78-94 pro 100.000 Personen geschätzt, oder grob 1 zu 1000 [17, 18]. Makroadenome machen einen signifikanten Anteil dieser klinisch relevanten Tumoren aus, was wahrscheinlich 30-50 % der diagnostizierten Adenome entspricht, insbesondere unter inaktiven Adenomen, die sich aufgrund ihrer Größe präsentieren.
Bevölkerungsbezogene Studien legen eine gesamte jährliche Inzidenz neu diagnostizierter Hypophysenadenome von rund 4 pro 100.000 Einwohner nahe [18], wobei die Prävalenzraten aufgrund verbesserter Erkennung durch Bildgebung steigen. Die Inzidenz, die einen neurochirurgischen Eingriff erfordert, liegt im Bereich von etwa 1 bis 7 pro 100.000 pro Jahr, wobei Makroadenome, die Masseneffekte oder Hormonüberschüsse verursachen, die auf eine medikamentöse Therapie nicht ansprechen, die primären Indikationen für eine Operation sind.
Morbidität und Mortalität bei Hypophysen-Makroadenomen
Die mit Hypophysen-Makroadenomen assoziierte Morbidität resultiert in erster Linie aus Masseneffekten (Sehverlust, Hirnnervenlähmungen, schwächende Kopfschmerzen), hormoneller Dysfunktion (Hypopituitarismus, der einen lebenslangen Ersatz erfordert, oder die systemischen Folgen von Hormonüberschusssyndromen wie Akromegalie oder Morbus Cushing) und potenziellen Komplikationen der Behandlung (z.B. chirurgische Risiken wie Liquorleck, Meningitis, Gefäßverletzung, Verschlechterung des Hypopituitarismus; Langzeiteffekte der Bestrahlung wie Hypopituitarismus, Sekundärtumoren, kognitive Veränderungen). Die Sehbehinderung, insbesondere permanente Defizite durch verzögerte Behandlung der Chiasmakompression, beeinträchtigt die Lebensqualität erheblich. Hypopituitarismus erfordert sorgfältiges Management und birgt eigene Risiken, wenn er unzureichend behandelt wird (z.B. Nebennierenkrise). Komplikationen von unkontrolliertem Hormonüberschuss (z.B. Herz-Kreislauf-Erkrankungen, Diabetes, Osteoporose, erhöhtes Krebsrisiko bei Akromegalie und Cushing) tragen signifikant zur langfristigen Morbidität und Mortalität bei, wenn sie nicht effektiv gemanagt werden [19, 20].
Die direkt mit der gutartigen Natur von Hypophysen-Makroadenomen selbst verbundene Mortalität ist im Allgemeinen gering. Ein erhöhtes Mortalitätsrisiko ist jedoch bei Patienten mit unkontrollierter Akromegalie und Morbus Cushing gut dokumentiert, hauptsächlich aufgrund von kardiovaskulären Komplikationen (Bluthochdruck, Herzinsuffizienz, Schlaganfall, Arrhythmien), Stoffwechselproblemen (Diabetes) und Infektionen (Cushing). Ein Hypophysenapoplex ist zwar selten, kann aber tödlich sein, wenn er nicht rechtzeitig erkannt und behandelt wird. Komplikationen durch Operationen (obwohl die Mortalität gering ist, <1%, in erfahrenen Zentren) oder Langzeiteffekte von Bestrahlung können ebenfalls beitragen. Das Gesamtüberleben für Patienten mit Makroadenomen ist im Allgemeinen gut mit modernem multidisziplinärem Management, das Hormonspiegel und Masseneffekt kontrolliert, obwohl die Lebensqualität durch Restsymptome oder Behandlungsnebenwirkungen beeinträchtigt sein kann.
Hypophysen-Makroadenome zeigen keine signifikante rassische Vorliebe.
Während Autopsiestudien eine gleiche Prävalenz von inzidentellen Adenomen zwischen den Geschlechtern zeigen, sind klinisch diagnostizierte Prolaktinome und ACTH-sezernierende Adenome (Morbus Cushing) bei Frauen im reproduktiven Alter signifikant häufiger, möglicherweise aufgrund früherer Symptomerkennung (Menstruationsstörungen, Galaktorrhoe). GH-sezernierende Adenome (Akromegalie) und klinisch präsentierende NFPAs weisen eine gleichmäßigere Geschlechterverteilung auf. Hypophysen-Makroadenome können in jedem Alter auftreten, werden aber am häufigsten bei Erwachsenen diagnostiziert, mit einem Höhepunkt der Inzidenz zwischen dem dritten und sechsten Lebensjahrzehnt.
Inaktive Hypophysenadenome (NFPAs)
Inaktive Hypophysenadenome (NFPAs) sind Hypophysentumore, die kein klinisches Syndrom der Hormonübersekretion verursachen. Sie machen etwa 15-30 Prozent aller klinisch diagnostizierten Hypophysenadenome aus und sind die häufigste Form, die sich als Makroadenome präsentiert [22]. Obwohl sie als "inaktiv" (nicht-funktionierend) bezeichnet werden, zeigt die immunhistochemische Färbung, dass die meisten NFPAs aus gonadotropen Zellen stammen und häufig Gonadotropine (Follikelstimulierendes Hormon - FSH, Luteinisierendes Hormon - LH) oder deren Untereinheiten (insbesondere die Alpha-Untereinheit oder FSH-beta-Untereinheit) produzieren. Diese Hormone sind jedoch typischerweise biologisch inaktiv, werden ineffizient oder in abnormen Verhältnissen sezerniert und verursachen daher keine klinischen Symptome eines Hormonüberschusses [6]. Einige können echte "Nullzell"-Adenome oder stumme Varianten anderer hormonproduzierender Zellen sein.
Da es bei ihnen keine Hormonhypersekretions-Syndrome gibt, präsentieren sich NFPAs gewöhnlich später als aktive Adenome, oft erst dann, wenn sie groß genug geworden sind (Makroadenome), um Symptome aufgrund des Masseneffekts zu verursachen:
- Gesichtsfeldausfälle (typischerweise bitemporale Hemianopsie durch Kompression des Chiasma opticum).
- Kopfschmerzen (unspezifisch, aufgrund von Dehnung der Dura oder sellärem Druck).
- Hypopituitarismus (aufgrund von Kompression des normalen Hypophysengewebes, wobei am häufigsten zuerst die GH- und Gonadotropinsekretion betroffen ist, gefolgt von TSH und ACTH).
- Hirnnervenlähmungen (seltener, durch Invasion in den Sinus cavernosus, verursacht Diplopie oder Gesichtstaubheit).
- Hypophysenapoplex (akute Blutung/Infarkt innerhalb des Tumors).
Viele kleinere NFPAs werden zufällig (MRT oder CT) als Hypophysen-Inzidentalome entdeckt. Autopsie- und Bildgebungsstudien weisen auf eine hohe Prävalenz von stummen Mikroadenomen hin (bis zu 1 von 6 Personen könnte möglicherweise eines haben), aber die meisten bleiben klein und asymptomatisch ein Leben lang.
Die primäre Behandlung für symptomatische NFPAs, die Masseneffekte oder einen progressiven Hypopituitarismus verursachen, ist die chirurgische Resektion, gewöhnlich über den transsphenoidalen Zugang. Die Hauptziele sind die Dekompression benachbarter Strukturen (insbesondere des optischen Apparats zur Verbesserung/Erhaltung der Sehkraft) und die maximale sichere Tumorentfernung zur Verringerung des Masseneffekts und zur potenziellen Verbesserung der Hypophysenfunktion. Das Management asymptomatischer inzidenteller NFPAs umfasst die Überwachung mit serieller Bildgebung und endokrinologischen/visuellen Beurteilungen [23].
Hypophysenadenom und Hyperprolaktinämie-Syndrom Diagnose
Differentialdiagnose von sellären/suprasellären Raumforderungen
| Bedingung | Hauptmerkmale / Unterscheidungsmerkmale | Typische Bildgebungsbefunde (MRT) |
|---|---|---|
| Hypophysenadenom (Mikro/Makro) | Endokrine Symptome (Überschuss/Mangel) oder Masseneffekt (Sehverlust, Kopfschmerzen, HN-Lähmung). Entsteht innerhalb der Sella, verdrängt die Drüse, kann sich supra/parasellär ausdehnen. | Mikro: Fokale Läsion <10mm, typischerweise T1-hypointens, Kontrastmittelaufnahme geringer/später als normale Drüse. Makro: Selläre Masse ≥10mm, variable KM-Aufnahme (oft geringer als Drüse), kann Blutung/Zyste zeigen, dehnt sich supra/parasellär aus, kann Knochen erodieren. |
| Kraniopharyngeom | Bimodaler Altersgipfel (Kinder/ältere Erwachsene). Supraselläre Lage häufig. Kopfschmerzen, Sehverlust, endokrine Dysfunktion (GH-Mangel, DI häufig), Hydrozephalus. | Oft gemischt zystische (T1 variabel/T2 hyper) und feste Komponenten. Verkalkung sehr häufig (CT am besten). Nimmt KM auf (feste Teile, dicke Zystenwand). Typischerweise supraselläres Epizentrum, kann an umliegenden Strukturen haften. |
| Meningeom (Sellär/Suprasellär/Sinus Cavernosus) | Erwachsene, F>M typischerweise. Langsam wachsend, durabasiert (Planum sphenoidale, Tuberculum sellae, Diaphragma, Wand des Sinus cavernosus). Masseneffekt (Sehverlust, HN-Lähmungen), Kopfschmerzen, Anfälle. Selten signifikante endokrine Dysfunktion, es sei denn, sehr groß. | Oft isointens T1/T2, intensive homogene KM-Aufnahme nach Kontrast, charakteristisches "Dural-Tail"-Zeichen häufig. Kann Hyperostose (Knochenverdickung) verursachen oder Gefäße umhüllen. |
| Rathke-Zyste | Gutartige Epithelzyste (Überbleibsel der Rathke-Tasche), oft zufällig. Kann Kopfschmerzen, Sehstörungen oder Hypopituitarismus verursachen, wenn sie groß ist. Meist intraselläre oder intra/supraselläre Mittellinienlage. | Gut definierte, nicht-anreichernde Zyste (gewöhnlich). Signal variiert stark (T1 hyper/iso/hypo, T2 meist hyper) basierend auf dem Proteingehalt. Dünne, glatte Wand, meist keine feste Komponente oder KM-Aufnahme (kann einen dünnen Rand zeigen). Intra-zystischer Knoten (proteinreich/hämorrhagisch) möglich (<30%). |
| Hypophysenhyperplasie | Vergrößerung von normalem Drüsengewebe. Physiologisch (Schwangerschaft, Stillzeit, Pubertät) oder pathologisch (Reaktion auf Endorganversagen wie primäre Hypothyreose, primäre Nebennierenrindeninsuffizienz). | Diffus vergrößerte Drüse (>10-12mm Höhe möglich), nimmt KM meist homogen und intensiv auf. Sella kann sich symmetrisch vergrößern. Drüsenform bleibt erhalten (konkave obere Begrenzung geht verloren). Bildet sich zurück, wenn die zugrunde liegende Ursache behandelt wird. |
| Hypophysitis (Lymphozytär, Granulomatös, IgG4) | Entzündung von Hypophyse/Stiel. Kopfschmerzen, Sehstörungen, Hypopituitarismus (oft rasches Einsetzen, multiple Achsen), Diabetes insipidus (DI) häufig (besonders Stielbeteiligung). Lymphozytär oft postpartal/autoimmuner Kontext. | Oft symmetrische Vergrößerung und Verdickung von Drüse/Stiel, diffuse intensive KM-Aufnahme. Kann ein Adenom imitieren, aber oft eher infiltratives Erscheinungsbild des Stiels. Klinischer Kontext entscheidend (rasch einsetzender Hypopituitarismus/DI). |
| Metastase | Anamnese von Primärkrebs (Brust, Lunge am häufigsten). Rasch einsetzender Hypopituitarismus, DI häufig (Prädilektion für Hypophysenhinterlappen/Stiel). Kopfschmerzen, Sehverlust, HN-Lähmungen möglich. | KM-aufnehmende Läsion, oft infiltrativ, betrifft häufig den Hypophysenhinterlappen oder -stiel. Können multiple intrakranielle Läsionen sein. Suche nach primärer Malignität ist der Schlüssel. |
| Aneurysma (Cavernosus/Supraselläre ICA) | Kann selläre Raumforderung imitieren, besonders wenn teilweise thrombosiert. Kann HN-Lähmungen (III, IV, V, VI wegen Lage im Sinus cavernosus), Kopfschmerzen verursachen. Risiko der Ruptur (SAB). | MRT zeigt charakteristisches "Flow-Void" (wenn durchgängig), laminiertes Signal von Thrombus (falls vorhanden), KM-Aufnahme der Wand. MRA/CTA/DSA bestätigt Diagnose und skizziert Gefäßanatomie. |
| Chordom / Chondrosarkom | Seltene bösartige/lokal aggressive Schädelbasistumoren aus Chorda-Überresten (Chordom) oder Knorpel (Chondrosarkom). Kann sich von Clivus/Felsenbeinspitze in Sella/supraselläre Region ausdehnen. Verursachen oft extensive Knochenzerstörung, HN-Lähmungen. | CT zeigt Knochenzerstörung. MRT zeigt lokal destruktive, KM-aufnehmende Masse, oft T2-hyperintens (bes. Chordom aufgrund physalipherer Zellen). Lage typischerweise auf Clivus oder petroklivalen Übergang zentriert. |
| Germinom (Suprasellär) | Seltener Keimzelltumor, meist Kinder/junge Erwachsene. Klassische Trias: DI, Sehverlust, Hypopituitarismus. Kann gleichzeitig die Zirbeldrüsenregion betreffen ("duale Mittellinientumoren"). | Homogen KM-aufnehmende supraselläre Masse, betrifft oft Chiasma opticum, Stiel/Hypothalamus. Liquor-/Serum-Tumormarker (AFP, β-hCG) können erhöht sein (obwohl oft normal beim reinen Germinom). Hoch strahlensensibel. |
Die Diagnose umfasst oft die Magnetresonanztomographie (MRT) der Hypophyse, den Goldstandard zur Visualisierung von Adenomen (Mikroadenome, Makroadenome, einschließlich Prolaktinome), Beurteilung ihrer Größe, Ausdehnung, Beziehung zu umliegenden Strukturen wie dem Chiasma opticum und Sinus cavernosus, und Hilfe bei der Unterscheidung von anderen sellären Läsionen.
Der diagnostische Ansatz kombiniert klinische Bewertung, Hormontests und Bildgebung:
- Klinischer Verdacht: Basierend auf Symptomen von Hormonüberschuss (z.B. Akromegalie-Merkmale, cushingoidale Zeichen, Galaktorrhoe/Amenorrhoe), Masseneffekt (Sehveränderungen, Kopfschmerzen, Hirnnervenlähmung), Hypopituitarismus (Müdigkeit, Hypogonadismus, Kälteintoleranz, etc.) oder einem Zufallsbefund in der Bildgebung.
- Hormonelle Abklärung: Die Messung basaler Spiegel von Hormonen des Hypophysenvorderlappens und der entsprechenden Zielorgane ist essenziell zur Identifikation von Hormonüberschuss oder -mangel. Wichtige Tests umfassen:
- Prolaktin (PRL): Erhöhte Spiegel deuten auf eine Hyperprolaktinämie hin. Deutlich erhöhte Werte (>200-250 ng/mL oder mcg/L) weisen sehr auf ein Prolaktinom (insbesondere ein Makroadenom) hin. Mäßig erhöhte Werte (25-150 ng/mL) erfordern die Untersuchung auf andere Ursachen (Medikamente, Hypothyreose, Stiel-Effekt, etc.). Auf "Hook-Effekt" mit verdünnter Probe prüfen, falls ein Makroadenom vorliegt und PRL unerwartet niedrig/normal ist [24].
- Wachstumshormon (GH) / Insulin-ähnlicher Wachstumsfaktor 1 (IGF-1): Ein erhöhtes alters- und geschlechtsangepasstes IGF-1 ist der beste Screening-Test für Akromegalie. Die Diagnose wird bestätigt durch das Ausbleiben einer GH-Suppression (< 0,4 oder < 1 ng/mL je nach Assay) während eines oralen Glukosetoleranztests (oGTT). Basale GH-Spiegel sind aufgrund der Pulsatilität weniger zuverlässig.
- ACTH / Cortisol: Das Screening auf das Cushing-Syndrom umfasst Tests wie Mitternachts-Speichelcortisol, 24-Stunden-Urin-freies Cortisol (UFC) oder niedrigdosierten (1 mg) Übernacht-Dexamethason-Hemmtest. Bei positivem Befund helfen Plasma-ACTH-Spiegel, ACTH-abhängige (Hypophysen/ektopisch) von -unabhängigen (Nebennieren) Ursachen zu unterscheiden. Hochdosierte Dexamethason-Hemmtests und/oder CRH-Stimulationstests, manchmal kombiniert mit Sinus petrosus inferior-Sampling (IPSS), helfen, den hypophysären Ursprung (Morbus Cushing) zu bestätigen [4].
- TSH / Freies T4/T3: Ein erhöhtes oder unangemessen normales TSH mit erhöhtem freiem T4/T3 deutet auf ein seltenes TSH-om hin. Die Tests screenen auch auf primäre Hypothyreose (hohes TSH, niedriges FT4), eine mögliche Ursache der sekundären Hyperprolaktinämie.
- LH / FSH / Testosteron (Männer) / Östradiol (Frauen): Beurteilung der Gonadenfunktion. Niedrige Werte deuten auf Hypogonadismus (sekundär, wenn LH/FSH ebenfalls niedrig/normal). Erhöhte Alpha-Untereinheit bei gonadotropen Adenomen häufig.
- Bewertung auf Hypopituitarismus: Wenn Masseneffekt oder Symptome einen Mangel nahelegen, Test von basalem morgendlichem Cortisol und ACTH, TSH und freiem T4, LH/FSH und Testosteron/Östradiol (entsprechend), IGF-1 (für GH-Achse). Dynamische Stimulationstests (z.B. ACTH-Stimulationstest, Insulintoleranztest - Goldstandard, aber riskant, GHRH-Arginin-Test) können nötig sein, um subtile Defizite zu bestätigen. Bewertung auf Diabetes insipidus (DI), wenn Hypophysenhinterlappen betroffen (Polyurie, Polydipsie, Serum/Urin-Osmolalität).
- Bildgebung:
- Hypophysen-MRT: Mit und ohne Gadolinium-Kontrastmittel, unter Verwendung dedizierter dünner Schichten (≤3mm) durch die Sella in koronalen und sagittalen Ebenen. Dynamische kontrastverstärkte Sequenzen sind besonders nützlich zur Identifizierung von Mikroadenomen, die oft als fokale Areale verzögerter Kontrastmittelaufnahme im Vergleich zum schnell anreichernden normalen Hypophysenvorderlappen erscheinen. Die MRT definiert präzise Tumorgröße, Verhältnis zum Chiasma opticum, Invasion in den Sinus cavernosus (Knosp-Klassifikation) und Stielabweichung [25].
- CT-Scan: Weniger empfindlich für die Adenome selbst, aber besser geeignet zur Darstellung knöcherner Erosionen am Sellaboden oder den Processus clinoidei sowie für Verkalkungen (häufiger bei Kraniopharyngeomen). Nützlich, wenn die MRT kontraindiziert ist.
- Augenärztliche Untersuchung: Eine formale Gesichtsfeldprüfung (typischerweise automatisierte Perimetrie, z.B. Humphrey-Gesichtsfeld) ist zwingend erforderlich, wenn sich ein Adenom in der Nähe oder in Kompression des Chiasma opticum befindet (Makroadenome), auch wenn der Patient keine visuellen Symptome berichtet. Die Beurteilung der Sehschärfe und des Aussehens des Sehnervs (Funduskopie auf Blässe/Atrophie oder Papillenödem) ist ebenfalls Standard.
- Differentialdiagnose der Hyperprolaktinämie: Wenn ein erhöhtes Prolaktin festgestellt wird, schließen Sie systematisch andere Ursachen aus, bevor Sie ein Prolaktinom diagnostizieren, insbesondere wenn die Werte nur mäßig hoch sind (<150 ng/mL) oder die Bildgebung normal/uneindeutig ist. In Betracht ziehen:
- Physiologische Ursachen: Schwangerschaft, Stillzeit, Stress, Schlaf (Morgenwerte prüfen), Bewegung, Brustwarzenstimulation, Geschlechtsverkehr.
- Medikamente: Überprüfung aktueller Medikamente (Antipsychotika - insb. Risperidon, typische; Antidepressiva - SSRIs, TCAs; Antiemetika - Metoclopramid, Domperidon; Antihypertensiva - Verapamil, Methyldopa; Opioide; Östrogene). Absetzen des verdächtigen Medikaments nach Möglichkeit und erneute Überprüfung des PRL nach ~3 Tagen (oder längerer Halbwertszeit).
- Systemische Erkrankungen: Primäre Hypothyreose (TSH/FT4 überprüfen), chronische Nierenerkrankung (Kreatinin überprüfen), schwere Leberzirrhose, polyzystisches Ovarialsyndrom (PCOS), Brustwandläsionen (Herpes zoster, Operationsnarben, Piercings).
- Hypophysenstiel-Effekt: Kompression des Hypophysenstiels durch nicht-Prolaktin-sezernierende Tumoren (NFPAs, Kraniopharyngeome, Meningeome) oder entzündliche Läsionen unterbricht die Dopaminzufuhr vom Hypothalamus zu Laktotrophen, was zu einer leichten/mittleren Hyperprolaktinämie (gewöhnlich <150 ng/mL) führt. Bildgebung ist der Schlüssel.
- Makroprolaktinämie: Vorhandensein großer, biologisch weniger aktiver Prolaktinkomplexe; erfordert spezifische Labortests (PEG-Präzipitation); klinisch in der Regel unbedeutend.
Normale Serum-Prolaktinwerte variieren leicht je nach Labor und Assay, liegen aber im Allgemeinen bei (ungefähre Bereiche):
Patientengruppe |
Ungefährer normaler Prolaktinbereich (ng/mL oder mcg/L) |
(Ungefähre SI-Einheiten: mIU/L) |
|---|---|---|
Kinder (präpubertär) |
Variabel, im Allgemeinen < 15-20 | ~ < 300-400 |
Erwachsene Männer |
< 15-20 | ~ < 300-400 |
Nichtschwangere erwachsene Frauen |
< 25-30 | ~ < 500-600 |
Schwangere Frauen |
Deutlich erhöht, steigt während der gesamten Schwangerschaft (z.B. bis zu 200-400+ ng/mL) | ~ bis zu 4000-8000+ mIU/L |
Nach der Geburt (stillend) |
Erhöht, Werte schwanken beim Stillen, sinken allmählich, wenn nicht gestillt wird | Variabel, kann anfangs sehr hoch sein |
Der Umrechnungsfaktor ist ungefähr (1 ng/mL ≈ 21 mIU/L basierend auf WHO-Standards, variiert aber je nach Assay).

Hypophysenadenom und Hyperprolaktinämie-Syndrom Behandlung
Ziele bei der Behandlung von Hypophysenadenomen sind die Normalisierung des Hormonspiegels (falls im Überschuss), Linderung der Symptome durch Masseneffekte (Sehverlust, Kopfschmerzen, Hirnnervenlähmungen), Erhaltung oder Wiederherstellung der normalen Hypophysenfunktion sowie Verhinderung eines Tumorrezidivs. Der spezifische Ansatz hängt stark von Tumorart (aktiv vs. inaktiv), Größe (Mikro vs. Makro), dem sezernierten Hormon, Vorhandensein und Schweregrad des Masseneffekts, Patientenalter, Fortpflanzungswünschen und Begleiterkrankungen ab [26, 2, 4, 23].
- Prolaktinome (Hyperprolaktinämie):
- Medizinische Therapie (Erste Wahl): Dopaminagonisten (DAs) sind hochwirksam und die bevorzugte Erstbehandlung für praktisch alle Prolaktinome, einschließlich großer Makroadenome, die Gesichtsfeldausfälle verursachen (es sei denn, der Sehverlust ist schwer/rasch fortschreitend).
- Cabergolin: Im Allgemeinen bevorzugt aufgrund höherer Wirksamkeit bei der Normalisierung des Prolaktins und Tumorschrumpfung, längerer Halbwertszeit (ermöglicht ein- oder zweimal wöchentliche Dosierung) und besserer Verträglichkeit im Vergleich zu Bromocriptin.
- Bromocriptin: Älterer Wirkstoff, erfordert tägliche Dosierung, häufiger assoziiert mit Übelkeit/posturaler Hypotonie, hat aber ein längeres Sicherheitsprofil, besonders in der Schwangerschaft.
- Operation (Neurochirurgischer Eingriff - Transsphenoidale Operation): In erster Linie indiziert bei Patienten mit Unverträglichkeit oder Resistenz gegenüber Dopaminagonisten, jenen mit Hypophysenapoplex in einem Prolaktinom, in seltenen Fällen bei großen zystischen/hämorrhagischen Komponenten, die auf DAs nicht ansprechen, oder manchmal bei Frauen mit Kinderwunsch, die DAs nicht vertragen oder vermeiden möchten. Heilungsraten sind bei Mikroadenomen (~70-90%) höher als bei invasiven Makroadenomen (~30-50%).
- Strahlentherapie (Stereotaktische Radiochirurgie oder fraktionierte RT): Reserviert für seltene Fälle von invasiven/aggressiven Prolaktinomen, die resistent gegen maximale medizinische und chirurgische Therapie sind oder wiederkehren. Vorwiegend zur Tumorkontrolle eingesetzt, da die Wirkung auf den Prolaktinspiegel langsam und oft unvollständig ist; hohes Risiko für Hypopituitarismus.
- Medizinische Therapie (Erste Wahl): Dopaminagonisten (DAs) sind hochwirksam und die bevorzugte Erstbehandlung für praktisch alle Prolaktinome, einschließlich großer Makroadenome, die Gesichtsfeldausfälle verursachen (es sei denn, der Sehverlust ist schwer/rasch fortschreitend).
- Akromegalie (GH-sezernierende Adenome):
- Operation (Im Allgemeinen erste Wahl): Die transsphenoidale Operation ist die primäre Behandlung mit dem Ziel der vollständigen Resektion und biochemischen Heilung (Normalisierung des alters-/geschlechtsangepassten IGF-1 und Suppression von GH während des oGTT). Die Heilungsraten hängen von der Tumorgröße, Invasivität und der Erfahrung des Chirurgen ab.
- Medizinische Therapie: Wird bei anhaltender Erkrankung nach der Operation verwendet, bei Patienten, die für eine Operation nicht in Frage kommen, oder manchmal präoperativ, um die Operationsergebnisse zu verbessern. Die Optionen umfassen:
- Somatostatin-Rezeptor-Liganden (SRLs): Die erste Generation (Octreotid LAR, Lanreotid Autogel) normalisiert IGF-1/GH bei ~50-60% und kann Tumore schrumpfen. Die zweite Generation (Pasireotid LAR) kann wirksam sein, wenn SRLs der ersten Generation versagen, birgt jedoch ein höheres Risiko für Hyperglykämie.
- GH-Rezeptor-Antagonist (Pegvisomant): Hochwirksam bei der Normalisierung von IGF-1 (~80-90%) durch Blockade der GH-Wirkung am Rezeptor; verkleinert den Tumor nicht und erfordert die Überwachung der Leberfunktion und der Tumorgröße.
- Dopaminagonisten (Cabergolin): Weniger wirksam, normalisiert IGF-1 nur bei ~10-30%, hauptsächlich als Zusatztherapie eingesetzt, insbesondere wenn der Tumor auch Prolaktin sezerniert.
- Strahlentherapie (Stereotaktisch oder fraktioniert): Wirksam zur Kontrolle des Tumorwachstums und zur langsamen Senkung der GH/IGF-1-Spiegel bei Patienten mit verbleibender/rezidivierender Erkrankung, die nicht auf medizinische Therapie ansprechen. Hohes Risiko für verzögerten Hypopituitarismus.
- Morbus Cushing (ACTH-sezernierende Adenome):
- Operation (Erste Wahl): Die transsphenoidale selektive Adenomektomie ist die Behandlung der Wahl und strebt eine biochemische Remission an, während die normale Hypophysenfunktion erhalten bleibt. Remissionsraten liegen bei ~70-90% für Mikroadenome in erfahrenen Zentren [27].
- Medizinische Therapie: Wird präoperativ zur Kontrolle des schweren Hyperkortisolismus, postoperativ bei persistierender/rezidivierender Erkrankung in Erwartung der RT-Wirkung oder als Primärtherapie verwendet, falls die Operation fehlschlägt/kontraindiziert ist. Medikamente zielen auf die Nebennierensteroidogenese (Ketoconazol, Metyrapon, Etomidat, Osilodrostat), die hypophysäre ACTH-Sekretion (Pasireotid, Cabergolin) oder die Glukokortikoid-Rezeptor-Blockade (Mifepriston) ab.
- Strahlentherapie (Stereotaktisch oder fraktioniert): Wirksam bei persistierender/rezidivierender Erkrankung nach einer Operation, aber die biochemische Remission ist verzögert (Monate bis Jahre).
- Bilaterale Adrenalektomie: Bietet eine definitive Heilung des Hyperkortisolismus, wenn auf die Hypophyse gerichtete Therapien fehlschlagen, führt jedoch zu einer dauerhaften Nebenniereninsuffizienz, die lebenslang einen Ersatz von Glukokortikoiden/Mineralokortikoiden erfordert, und birgt das Risiko des Nelson-Syndroms (progressives Wachstum/Pigmentierung durch den hypophysären ACTH-sezernierenden Tumor).
- TSH-ome (selten):
- Operation (Erste Wahl): Die transsphenoidale Resektion ist die bevorzugte Behandlung.
- Medizinische Therapie: Somatostatin-Rezeptor-Liganden (Octreotid, Lanreotid) sind oft wirksam bei der Unterdrückung der TSH-Sekretion und Tumorschrumpfung präoperativ oder bei einer Resterkrankung. Antithyreoidale Medikamente (Methimazol, PTU) lindern Hyperthyreose-Symptome, behandeln aber nicht den Tumor.
- Strahlentherapie: Für residuelle/rezidivierende Tumore, die auf andere Therapien nicht ansprechen.
- Inaktive Hypophysenadenome (NFPAs): Siehe Abschnitt unten.
Managemententscheidungen sollten individuell getroffen werden und erfordern häufig einen multidisziplinären Teamansatz unter Einbeziehung von Endokrinologen, Neurochirurgen, Neuroradiologen, Augenärzten und manchmal Radioonkologen.
Behandlung inaktiver Hypophysenadenome (NFPAs)
Da NFPAs typischerweise keine Hormonüberschusssyndrome verursachen, ist die Behandlung vor allem dann indiziert, wenn sie Masseneffekte (Sehbehinderung, Hirnnervenlähmungen, klinisch signifikante, auf den Tumor zurückzuführende Kopfschmerzen) oder Hypopituitarismus verursachen, oder wenn sie ein signifikantes Wachstum in der seriellen Bildgebung zeigen [22, 23].
- Asymptomatische inzidentelle NFPAs:
- Mikroadenome (<10mm): Im Allgemeinen konservativ behandelt mit regelmäßiger MRT-Überwachung (z. B. MRT nach 1 Jahr, dann 1-2 Jahre, dann seltener, falls stabil) und grundlegender endokriner Untersuchung (Screening auf Hypopituitarismus und Hyperprolaktinämie aufgrund eines Stieleffekts). Die meisten (~90-95%) wachsen nicht signifikant [8].
- Makroadenome (≥10mm) ohne Masseneffekt (Keine visuellen Defizite, normale Hypophysenfunktion, nicht an das Chiasma angrenzend): Das Management erfolgt individuell. Die Optionen umfassen eine engmaschige Überwachung mit MRT (z. B. nach 6 Monaten, dann jährlich, falls stabil) und regelmäßige endokrine/visuelle Beurteilungen, oder die Erwägung einer Operation, insbesondere wenn der Tumor groß oder sehr nah am Chiasma opticum ist, angesichts des Potenzials für zukünftiges Wachstum/Komplikationen. Ein Eingriff ist indiziert bei dokumentiertem Tumorwachstum, Entwicklung von Symptomen (Sehverlust, Hypopituitarismus) oder wenn der Tumor das Chiasma opticum berührt/komprimiert [23].
- Symptomatische NFPAs (verursachen Masseneffekt oder Hypopituitarismus):
- Chirurgischer Eingriff (Erste Wahl): Transsphenoidale Operation ist die primäre Behandlung. Die Hauptziele sind die Dekompression des optischen Apparats zur Verbesserung/Erhaltung der Sicht und die maximale sichere Tumorentfernung zur Linderung des Masseneffekts und zur potenziellen Erholung der Hypophysenfunktion (weniger wahrscheinlich, wenn die Defizite langanhaltend sind). Endoskopische endonasale Zugänge werden heutzutage häufig verwendet. Eine vollständige Resektion ist oft schwierig bei großen oder invasiven (z.B. in den Sinus cavernosus) Tumoren.
- Strahlentherapie (Stereotaktische Radiochirurgie - SRS oder fraktionierte RT): In erster Linie indiziert als adjuvante Behandlung bei erheblichem Resttumor nach einer Operation (insbesondere wenn es sich um ein wachsendes oder großes Restvolumen handelt) oder bei rezidivierenden Tumoren. Primäre RT wird selten verwendet, es sei denn, eine Operation ist kontraindiziert. RT bietet exzellente langfristige Tumorkontrollraten (>90%), reduziert jedoch den Masseneffekt nicht sofort und birgt ein signifikantes verzögertes Risiko (~30-60% über 10 Jahre) für strahleninduzierten Hypopituitarismus [28].
- Medizinische Therapie: Es gibt derzeit keine wirksame medizinische Therapie, die NFPAs konsistent schrumpft oder ihr Wachstum verhindert. Dopaminagonisten oder Somatostatinanaloga wurden untersucht, sind jedoch bei typischen gonadotropen NFPAs im Allgemeinen unwirksam.
- Management von Hypopituitarismus: Hormondefizite, die vor oder nach der Behandlung festgestellt werden, erfordern eine angemessene und lebenslange physiologische Ersatztherapie (z.B. Hydrocortison bei Nebenniereninsuffizienz - Stressdosisabdeckung erforderlich; Levothyroxin bei sekundärer Hypothyreose; Testosteron/Östrogen +/- Progesteron bei Hypogonadismus; potenziell GH-Ersatz bei Erwachsenen mit bestätigtem Mangel und signifikanten Auswirkungen auf die Lebensqualität oder Stoffwechselparameter; DDAVP bei Diabetes insipidus, falls vorhanden).
- Langfristige Nachsorge: Unerlässlich nach Behandlung (Operation +/- Bestrahlung) für alle Makroadenome. Umfasst die Überwachung auf Tumorrezidiv oder erneutes Wachstum (serielle MRT, Frequenz hängt von Resttumor/Behandlungsart ab), die regelmäßige Beurteilung der endokrinen Funktion (jährliche Tests auf Hypopituitarismus, auch nach RT) und die Überwachung des visuellen Status (Gesichtsfelder bei Vorgeschichte von Kompression). Die Rezidivraten nach alleiniger Operation können im Laufe der Zeit signifikant sein (10-50% je nach Nachbeobachtungszeit und Resttumor), was die Erwägung einer adjuvanten Strahlentherapie für bedenkliche Resttumoren notwendig macht.
![]() Achtung! Hypophysenadenome erfordern eine spezialisierte Diagnose und ein Management durch ein multidisziplinäres Team. Symptome wie Sehveränderungen, anhaltende Kopfschmerzen, hormonelle Ungleichgewichte (Menstruationsveränderungen, Laktation, unerklärte körperliche Veränderungen, sexuelle Dysfunktion) oder übermäßige Müdigkeit rechtfertigen die Untersuchung durch einen Arzt, die möglicherweise zur Überweisung an einen Endokrinologen und Neurochirurgen führt.
Achtung! Hypophysenadenome erfordern eine spezialisierte Diagnose und ein Management durch ein multidisziplinäres Team. Symptome wie Sehveränderungen, anhaltende Kopfschmerzen, hormonelle Ungleichgewichte (Menstruationsveränderungen, Laktation, unerklärte körperliche Veränderungen, sexuelle Dysfunktion) oder übermäßige Müdigkeit rechtfertigen die Untersuchung durch einen Arzt, die möglicherweise zur Überweisung an einen Endokrinologen und Neurochirurgen führt.
Referenzen
- Melmed S. Pituitary-Tumor Endocrinopathies. N Engl J Med. 2020 Mar 5;382(10):937-950. doi: 10.1056/NEJMra1810772
- Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and Treatment of Hyperprolactinemia: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2011 Feb;96(2):273-88. doi: 10.1210/jc.2010-1692
- Katznelson L, Laws ER Jr, Melmed S, et al; Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Nov;99(11):3933-51. doi: 10.1210/jc.2014-2700
- Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008 May;93(5):1526-40. doi: 10.1210/jc.2008-0125
- Beck-Peccoz P, Persani L. Thyrotropin-secreting pituitary adenomas. N Engl J Med. 1996;334(11):712-713. doi: 10.1056/NEJM199603143341107
- Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2011 Jun;14(2):111-8. doi: 10.1007/s11102-011-0300-1
- Molitch ME. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 1997 Mar;26(1):1-17. doi: 10.1016/s0889-8529(05)70239-7
- Fernández-Balsells MM, Murad MH, Barwise A, et al. Clinical management of nonfunctioning pituitary adenomas (NFPAs): a systematic review and meta-analysis. J Clin Endocrinol Metab. 2011 Apr;96(4):905-15. doi: 10.1210/jc.2010-1921
- Chapter on Pituitary Imaging. In: Osborn AG, Salzman KL, Jhaveri MD, et al. Osborn's Brain. 2nd ed. Elsevier; 2018.
- Asa SL, Ezzat S. The pathogenesis of pituitary tumours. Nat Rev Cancer. 2002 Feb;2(2):83-94. doi: 10.1038/nrc760
- Vierimaa O, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006 May 26;312(5777):1228-30. doi: 10.1126/science.1126100
- Reincke M, et al. Mutations in the deubiquitinase gene USP8 cause Cushing's disease. Nat Genet. 2015 Jan;47(1):31-8. doi: 10.1038/ng.3166
- Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936 Mar;12(2):205-16.1.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004 Aug 1;101(3):613-9. doi: 10.1002/cncr.20412
- Hall WA, et al. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994 May 15;120(10):817-20. doi: 10.7326/0003-4819-120-10-199405150-00001
- Ciccarelli A, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Varese, Italy. J Clin Endocrinol Metab. 2001 Feb;86(2):720-5. doi: 10.1210/jcem.86.2.7204
- Fernandez A, et al. Prevalence of pituitary adenomas: a community-based study in Olmsted County, Minnesota, 1993-1997. Arch Intern Med. 2000 Jan 10;160(1):78-81. doi: 10.1001/archinte.160.1.78
- Daly AF, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006 Dec;91(12):4756-69. doi: 10.1210/jc.2006-1668
- Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009 Mar;119(3):429-38. doi: 10.1172/JCI38090
- Nieman LK. Cushing's syndrome: update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015 Oct;173(4):M33-8. doi: 10.1530/EJE-15-0464
- Elster AD, et al. Pituitary gland: MR imaging of physiologic hypertrophy in adolescence. Radiology. 1990;174(3 Pt 1):681-5. doi: 10.1148/radiology.174.3.2305044
- Molitch ME. Nonfunctioning pituitary adenomas. Pituitary. 2005;8(3-4):213-20. doi: 10.1007/s11102-006-8512-6
- Freda PU, Beckers AM, Katznelson L, et al; Endocrine Society. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011 Apr;96(4):894-904. doi: 10.1210/jc.2010-1048
- Frieze TW, et al. Hook effect in prolactinomas: case report and review of literature. Endocr Pract. 2006;12(3):296-303. doi: 10.4158/EP.12.3.296
- Bonneville F, et al. Radiology of pituitary adenomas: an update. Radiol Clin North Am. 2012 Nov;50(6):995-1013. doi: 10.1016/j.rcl.2012.08.005
- Melmed S, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509-17. doi: 10.1210/jc.2008-2421
- Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing's Syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015 Aug;100(8):2807-31. doi: 10.1210/jc.2015-1818
- Sheehan JP, et al. Radiosurgery for nonfunctioning pituitary adenomas: a systematic review. J Neurosurg. 2013 Aug;119(2):446-56. doi: 10.3171/2013.4.JNS121833
Siehe auch
- Anatomie des Nervensystems
- Infektionen des Zentralnervensystems:
- Hirnabszess (lobär, zerebellär)
- Eosinophiles Granulom, Langerhans-Zell-Histiozytose (LCH), Hennebert-Symptom
- Epiduraler Hirnabszess
- Sinusitis-assoziierte intrakranielle Komplikationen
- Otogene intrakranielle Komplikationen
- Sinusitis-assoziierte ophthalmologische Komplikationen
- Bakterielle otogene Meningitis
- Subduraler Hirnabszess
- Eitrige Thrombophlebitis des Sinus sigmoideus
- Kolloidzyste des 3. Ventrikels
- Zerebrale und spinale adhäsive Arachnoiditis
- Kortikobasale Degeneration (Limitierte Hirnatrophie)
- Enzephalopathie
- Kopfschmerzen, Migräne
- Schädel-Hirn-Trauma (Gehirnerschütterung, Prellung, Hirnblutung, axonale Scherverletzungen)
- Erhöhter Hirndruck und Hydrozephalus
- Parkinson-Krankheit
- Hypophysen-Mikroadenom, Makroadenom und inaktive Adenome (NFPAs), Hyperprolaktinämie-Syndrom
- Spontaner kranialer Liquorleck (Liquorrhoe)